Commission, January 10, 2020, No M.9461
EUROPEAN COMMISSION
Decision
ABBVIE / ALLERGAN
To the notifying party
Subject: Case M.9461 – ABBVIE / ALLERGAN Commission decision pursuant to Article 6(1)(b) in conjunction with Article 6(2) of Council Regulation No 139/20041 and Article 57 of the Agreement on the European Economic Area2
Dear Sir or Madam,
(1) On 12 November 2019, the European Commission received notification of a proposed concentration pursuant to Article 4 of the Merger Regulation (the “Transaction”) by which AbbVie Inc. (“AbbVie”, United States), acquires within the meaning of Article 3(1)(b) of the Merger Regulation control of the whole of Allergan plc (“Allergan”, Ireland, and together with AbbVie the “Parties”).
1. THE PARTIES AND THE OPERATION
(2) AbbVie is a global pharmaceutical company listed on the New York Stock Exchange and headquartered in the United States. AbbVie is engaged in the development and commercialisation of innovative medicines in six main therapeutic areas: immunology (including autoimmune diseases), oncology, virology, neuroscience/central nervous system disorders, metabolic diseases and pain associated with endometriosis.
(3) Allergan is a global pharmaceutical company listed on the New York Stock Exchange and headquartered in Ireland. Allergan is engaged in the development and commercialisation of medicines in four main therapeutic areas: medical aesthetics, eye care, neuroscience/central nervous system disorders and gastroenterology.
(4) On 25 June 2019, AbbVie and Allergan signed an agreement (the “Transaction Agreement”). Under the terms of the Transaction Agreement, upon closing of the proposed Transaction, AbbVie will acquire 100% of the shares and therefore sole control of Allergan, in a cash and stock transaction. The Transaction would therefore give rise to a concentration within the meaning of Article 3(1)(b) of the Merger Regulation.
2. EU DIMENSION
(5) The undertakings concerned have a combined aggregate world-wide turnover of more than EUR 5 000 million3. Each of them has an EU-wide turnover in excess of EUR 250 million, but each of them does not achieve more than two-thirds of its aggregate EU-wide turnover within one and the same Member State. The notified operation therefore has an EU dimension.
3. FRAMEWORK FOR THE COMMISSION’S COMPETITIVE ASSESSMENT
3.1. General considerations on market definition in the pharmaceutical sector
3.1.1. Relevant product market
(6) When defining relevant markets in past decisions dealing with finished dose pharmaceutical products (“FDPs”),4 the Commission based its assessment on the following general approach.5
(7) The ATC system is a hierarchical and coded four-level system, which classifies medicinal products by class according to their indication, therapeutic use, composition, and mode of action (“MoA”). In the first and broadest level (ATC 1), medicinal products are divided into the 16 anatomical main groups. The second level (ATC 2) is either a pharmacological or therapeutic group. The third level (ATC 3) further groups medicinal products by their specific therapeutic indications. Finally, the ATC 4 level is generally the most detailed one (not available for all ATC 3) and refers for instance to the MoA or any other subdivision of the relevant products.
(8) The Commission has referred to the third level (ATC 3) as the starting point for defining the relevant product market. However, in several cases, the Commission found that the ATC 3 level classification did not yield the appropriate market definition within the meaning of the Commission Notice on the Definition of the Relevant Market.6 In particular, the Commission has considered in previous decisions plausible product markets at the ATC 4 level, at a level of a molecule or a group of molecules considered interchangeable and exercising competitive pressure on one another.7
(9) The Commission has also envisaged the possibility of defining the market by reference to the disease treated (and its degree of severity). For instance, in oncology, the Commission took into consideration the type of cancer, its location and whether the cancer is in an initial or an advanced stage.8 Similarly, in autoimmune diseases, the Commission has typically identified relevant product markets by reference to indications.9
(10) In its past decisional practice, the Commission has also considered relevant market segmentations based on (i) the types of treatment (e.g. chemotherapy, targeted therapies and immunotherapies in oncology;10 conventional and biologic treatments in autoimmune diseases),11 (ii) the line of treatment,12 (iii) the MoA,13 and (iv) the mode of delivery (“MoD”, e.g. oral, intravenous, intramuscular, and subcutaneous injections).14
(11) As regards pharmaceutical products in development (also called pipeline products), the Commission has in previous decisions considered market definitions based on the indication, the MoA, and, where relevant, the line of treatment, but ultimately left open the exact delineation of the market.15 The Commission added that when research and development (“R&D”) activities are assessed in terms of importance for future markets, the product market definition can be less clearly defined than for marketed products, reflecting the intrinsic uncertainty in analysing products that do not exist yet.16
(12) The Commission will analyse in Section 4 below the relevance of these distinctions for the relevant product market definition in the present case.
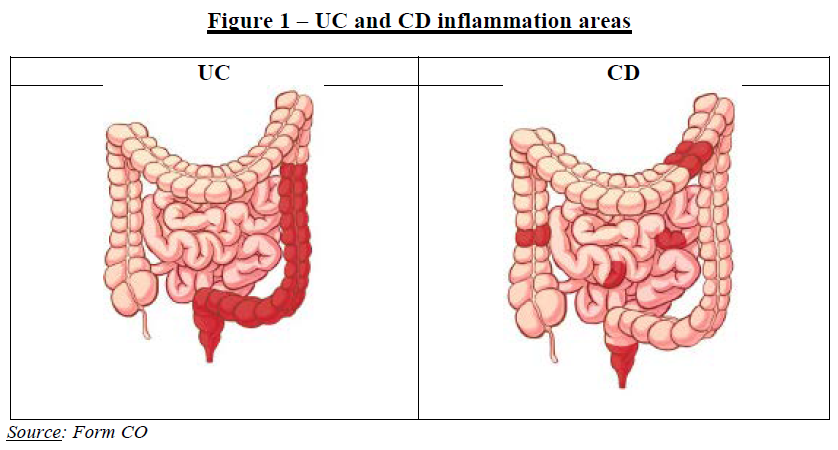
3.1.2. Relevant geographic market
(13) The Commission has consistently considered that the markets for FDPs are national in scope, in particular in view of the national regulatory and reimbursement schemes and the fact that competition between pharmaceutical firms still predominantly takes place at a national level.17 For pipeline products, the Commission has considered the geographic scope of the market to be at least EEA-wide.18
(14) The Commission will analyse in Section 4 below the relevance of these precedents for the relevant geographic market definition in the present case.
3.2. General approach to competitive assessment of horizontal effects of the Transaction
(15) Article 2 of the Merger Regulation requires the Commission to examine whether notified concentrations are compatible with the internal market, by assessing whether they would significantly impede effective competition in the internal market or in a substantial part of it, in particular, as a result of the creation or strengthening of a dominant position or the removal of a significant competitive constraint.
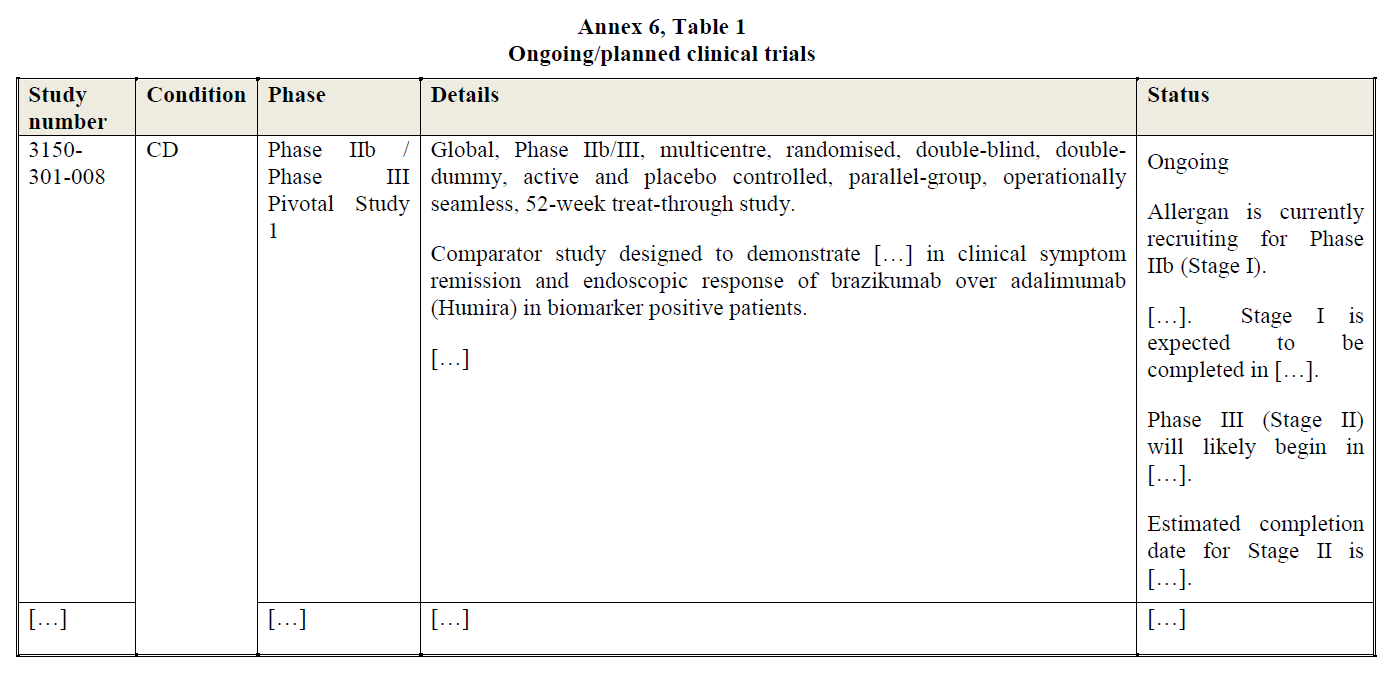
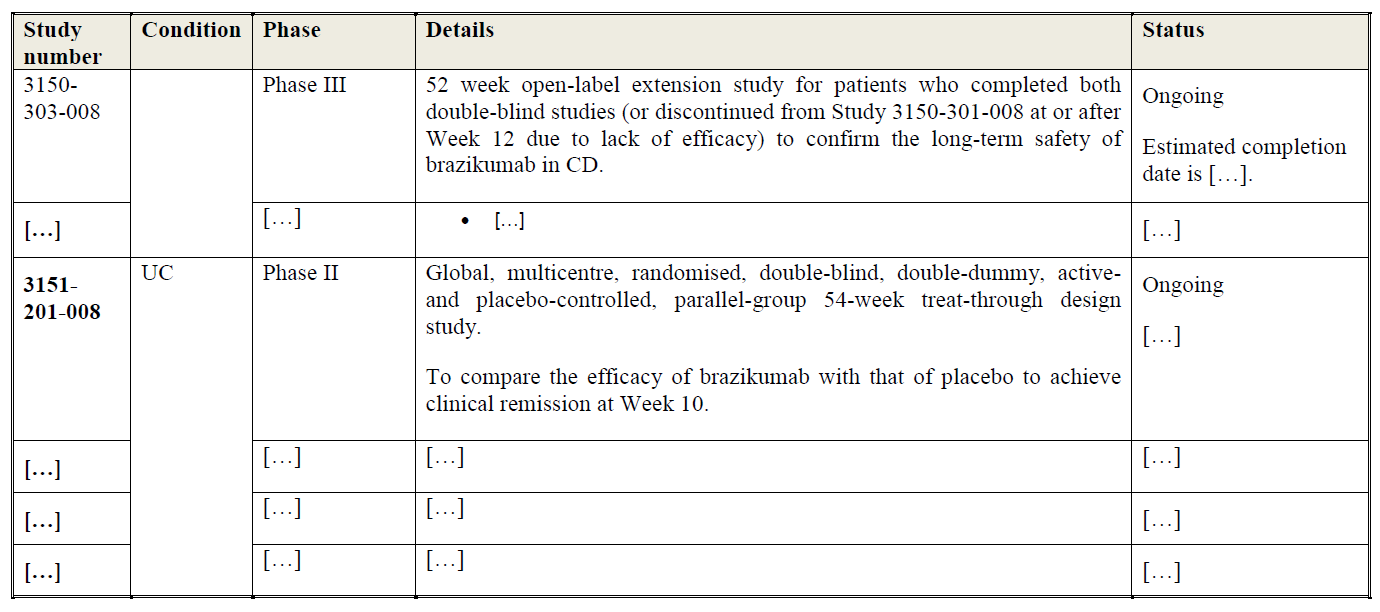
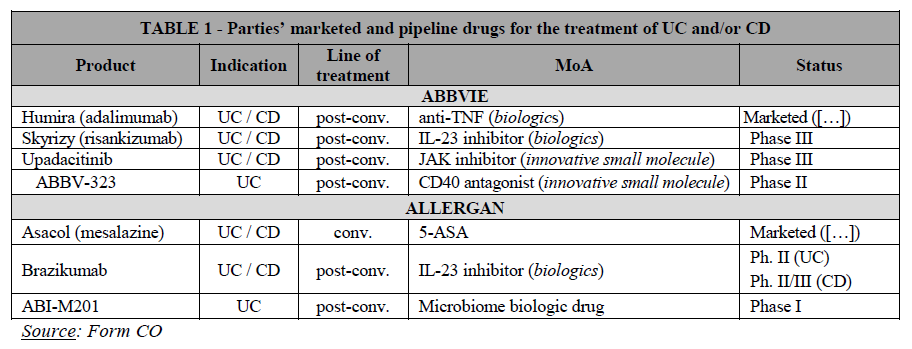
(16) In addition, Article 57(1) of the EEA Agreement requires the Commission to examine whether notified concentrations are compatible with the functioning of the EEA Agreement, by assessing whether they would create or strengthen a dominant position as a result of which effective competition would be significantly impeded within the EEA territory or a substantial part of it.
(17) In this framework, “competition” is understood to mean product and price competition (actual or potential), as well as innovation competition, where the Commission assesses in particular potential horizontal non-coordinated effects.19 The Commission considers that a concentration may not only affect competition in existing markets, but also competition in innovation and new product markets.20 This may be the case when a concentration concerns entities currently developing new products or technologies which may one day replace existing ones or which are being developed for a new intended use and will therefore not replace existing products but create a completely new demand.21
(18) In the pharmaceutical industry, the process of innovation is structured in such a way that it is typically possible to identify competing research programmes (or “pipeline” programmes) at an early stage of clinical trials.22 Competing pipeline programmes can be defined as R&D efforts aimed at developing substitutable products and having similar timing. The timing of a research programme should be assessed by reference to the stage of the on-going preclinical or clinical trials.23
(19) In line with the past decisional practice in the pharmaceutical sector24 and the Commission’s decisions in Dow/Dupont and Bayer/Monsanto,25 the Commission has taken into account a four-layer competitive assessment framework, which corresponds to the overlaps between the Parties’ activities in terms of:
(a) Actual (product and price) competition, assessing the overlaps between the Parties' existing (marketed) products;
(b) Potential (product and price) competition, assessing the overlaps (i) between the parties’ existing (marketed) and pipeline products at advanced stages of development and (ii) between the parties’ pipeline products at advanced stages of development. For pharmaceutical products, the Commission in principle considers programmes in Phase II and III clinical trials as being at an advanced stage of development;26
(c) Innovation competition in relation to the parties’ ongoing pipeline products, assessing the risk of significant loss of innovation competition resulting from the discontinuation, delay or redirection of the overlapping pipelines (including early stage pipelines); and
(d) Innovation competition in relation to the capability to innovate in certain innovation spaces, assessing the risk of a significant loss of innovation competition resulting from a structural reduction of the overall level of innovation.27
(20) The Commission will analyse the overlaps between the activities of the Parties against this framework in Section 4 below.
4. COMPETITIVE ASSESSMENT
(21) AbbVie and Allergan are both active in the development and commercialisation of pharmaceutical products. The Parties’ activities are highly complementary and only give rise to limited actual horizontal overlap in the EEA in relation to marketed and/or pipeline treatments in (i) inflammatory bowel diseases (“IBD”), covering ulcerative colitis (“UC”) and Crohn’s disease (“CD”), as well as (ii) uveitis.28
4.1. Treatments for UC and CD
4.1.1. Introduction
4.1.1.1. Overview of the diseases
(22) UC is a chronic autoimmune disease that causes inflammation and ulceration of the inner superficial lining of the large intestine. Inflammation usually begins in the rectum and lower colon, but may affect the entire colon.
(23) CD is a chronic autoimmune disease that affects any part of the gastrointestinal tract from the mouth to the rectum, affecting the full thickness of the intestinal wall (the superficial lining as well as deeper within the bowel wall). Most commonly, CD affects the last part of the small intestines, or the colon. The area of inflammation may be small (only a few centimetres) or may extend quite a distance along part of the bowels.
(24) The main difference between UC and CD is the areas affected by each disease. A schematic overview of the areas affected by UC and CD is provided below:
(25) Symptoms experienced by patients suffering from UC and CD are similar and include abdominal pain, severe diarrhoea, fatigue, malnutrition, severe weight loss and frequent bowel movements.
(26) The European Crohn’s and Colitis Organisation (“ECCO”) sets out (separate) guidelines for the treatment of UC and CD. The ECCO guidelines distinguish between mild, moderate or severe UC and CD based on a number of factors including (i) number of bowel movements and the presence of blood in stools, as well as temperature, pulse, haemoglobin, and C-reactive protein (“CRP”) levels for UC; (ii) a Crohn’s Disease Activity Index (“CDAI”), weight loss, fever, and CRP levels for CD.
4.1.1.2. Treatment algorithm
(27) The treatment for UC and CD follow sequences (or “algorithms”), meaning that the patient is initially treated with one type of drugs and moves on to another type if the initial drug does not work or stops working after a certain period of time. The algorithms for UC and CD are to a large extent similar and consist of:
- Conventional treatments, including aminosalicylates (“5-ASA”, typically mesalazine), corticosteroids and immunosuppressants;
- Post-conventional treatments (prescribed after the failure of conventional therapies or in case of contraindication), including biologic drugs and innovative small molecules. Biologic drugs include drugs with different MoAs, namely anti- Tumour Necrosis Factor (“anti-TNFs”), anti-integrins, interleurkin (“IL”)-12/23 inhibitors, and IL-23 inhibitors (currently under development). There is currently only one innovative small molecule approved and marketed in the EEA for the treatment of UC (not CD), marketed by Pfizer (Xeljanz), which was launched in the EEA in late 2018.
(28) UC and CD are both characterised by phases of remission and relapse. Patients may be switched back from post-conventional treatments to conventional treatments to maintain remission. Within post-conventional treatments, patients are also typically prescribed treatments in succession over the course of the disease. This is because a substantial proportion of UC and CD patients have an inadequate response or progressively lose response to a drug and need to switch to another treatment. As a result, there are also first, second, and/or third line post-conventional treatments.
4.1.2. The Parties’ activities
(29) The Parties’ marketed and pipeline drugs for the treatment of UC and/or CD are detailed in Table 1 below.
(30) […]. AbbVie markets a post-conventional treatment indicated for moderate-tosevere UC and CD in the EEA, namely Humira (adalimumab), an anti-TNF. AbbVie is also currently developing Skyrizi (risankizumab), an IL-23 inhibitor currently in Phase III clinical trials for both UC and CD (and approved for the treatment of psoriasis). In addition, AbbVie is also developing a JAK inhibitor (upadacitinib), which is in Phase III for both UC and CD, and a CD-40 antagonist, which is currently in Phase II for UC.
(31) Allergan markets a conventional treatment for mild-to-moderate UC and (to a lesser extent) CD […], namely Asacol-HD (mesalazine). Allergan does not currently market post-conventional treatments for UC or CD in the EEA. However, Allergan is developing brazikumab, an IL-23 inhibitor currently in Phase II clinical trial for UC and Phase II/III clinical trial for CD. Allergan is also developing ABI-M201, an early-stage (Phase I) pipeline for an orally administered microbiome biologic drug for the treatment of UC.
4.1.3. Market definition
4.1.3.1. Commission’s precedents
(32) The Commission has previously assessed the market for UC and CD treatments in several cases, including in particular in cases M.7379 – AbbVie/Shire and M.8955 – Takeda/Shire.29 In these decisions, the Commission considered a segmentation based on the line of treatment and found that conventional treatments (such as mesalazine) and post-conventional treatments (such as anti-TNFs) do not belong to the same product market.30
(33) Within post-conventional treatments, the Commission envisaged a segmentation based on MoA (e.g. anti-TNFs, anti-integrins, IL-12/23 inhibitors). In Takeda/Shire, the Commission found that anti-integrin biologics had a better safety profile than anti-TNFs and IL-12/23 inhibitors (which have a general immunosuppressant effect) and concluded, on this basis, and for the purpose of that decision, that anti-integrins constituted a distinct product market.31 Most recently, in case M.9294 - BMS/Celgene, the Commission considered potential segmentations of the IBD treatments market by line of treatment, MoA or MoD, but ultimately left the relevant product market definition open.32
4.1.3.2. The Parties’ views
(34) The Parties submit it is appropriate to consider a market for post-conventional treatments of moderate-to-severe UC and CD distinct from that of conventional therapies.
(35) In light of the Takeda/Shire decision, the Parties submit that a relevant market for post-conventional treatments of moderate to severe UC and CD would include all such treatments excluding anti-integrins, which the Commission held to be a separate market. This market would thus cover anti-TNFs, IL-inhibitors (including both IL-12/23 and IL-23 inhibitors), and innovative small molecule treatments (including JAK inhibitors and other products under development such as S1P receptor agonists).
4.1.3.3. The Commission’s assessment
(36) In line with the Commission’s precedents, the market investigation indicated that the treatments and the competitive dynamics are similar in UC and CD.33 Treatments for these two diseases are thus assessed jointly, and specificities are flagged where relevant.
(37) The market investigation confirmed that (i) conventional and (ii) post-conventional treatments for UC and CD belong to separate markets. This is due to the fact that conventional treatments are used to treat patients suffering from less severe forms of UC and CD,34 before the use of post-conventional biologics and innovative small molecule drugs.35
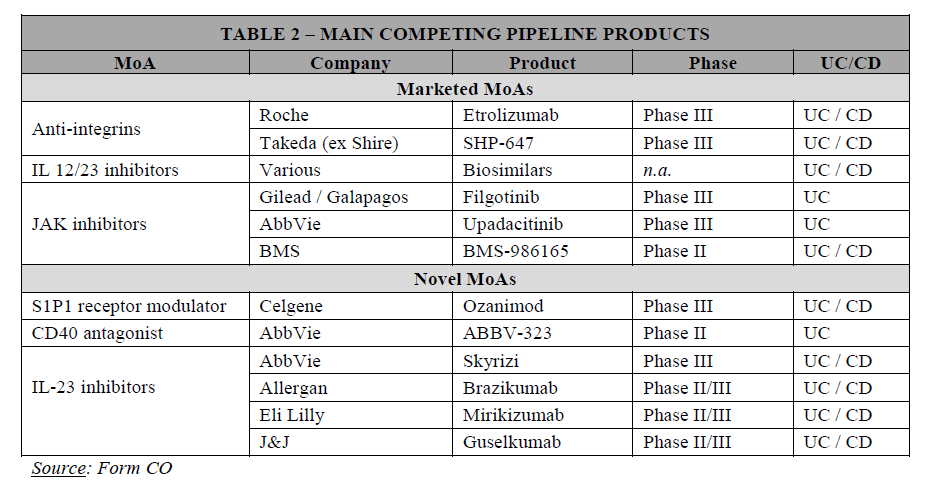
(38) The remainder of this Section focuses on the potential segmentations of the postconventional treatments for UC and CD. In this respect, the market investigation was not conclusive, in part because some of the drugs analysed (notably IL-23 inhibitors) are still at the development stage and there is an intrinsic level of uncertainty in assessing their future characteristics and market positioning.
(A) Potential segmentation of the post-conventional treatments market based on the line of treatment
(39) Based on the results of the market investigation, it is not clear whether a segmentation based on the line of treatment is warranted.
(40) On the one hand, the results of the market investigation confirmed that biologics and innovative small molecules are often prescribed in succession with anti-TNFs (first generation biologics).36
(41) The use of anti-TNFs as first-line post-conventional treatments is a requirement in some countries such as Austria and France due to reimbursement restrictions but also applies to other countries, where anti-TNFs are used in practice as first-line postconventional treatments for cost reasons (as anti-TNFs are cheaper in particular due to the availability of biosimilars).37 One competitor noted for instance that “[t]here are several EEA countries where the use of anti-TNF as a first–line postconventional treatment for UC/CD is encouraged and certain measures are in place. The level of these measures may vary however from obligatory use of anti-TNF to strong recommendation / incentivization of the use of the cheapest treatment in firstline post conventional UC/CD treatment populations. Different guidance may apply on national and regional (or hospital) level”. One Key Opinion Leader (“KOL”) also noted that “[g]enerally the first line biologic is anti-TNF – other biologics are used in specific instances. Failure of anti- TNF leads to initiation of other biologics or oral synthetic drug Tofacitinib”. 38
(42) This is also illustrated by the fact that a majority of customers expects that the launch of IL-23 inhibitors will have a more limited impact on the sale of anti-TNFs than on the sales of anti-integrins, IL-12/23 and JAK inhibitors, which are currently still mainly prescribed after the use of anti-TNFs.39 This is due to the fact that IL-23 inhibitors are also expected to be used as second or further line post-conventional treatments, at least at the time of their launch.40
(43) On the other hand, the market investigation revealed a certain degree of substitutability between post-conventional treatments for UC and CD.
(44) While anti-TNFs are currently the standard first-line post-conventional treatment for UC and CD, they are subject to competition from other treatments. A majority of responding competitors and KOLs consider that all post-conventional treatments compete with each other.41 One competitor noted for instance that “[i]n Immunology in general, and in GI [gastroenterology] specifically, [TNF inhibitors, IL-12/23 inhibitors, anti-integrin, and JAK inhibitors] compete with one another to a large extent”. Another one confirmed that “[d]ifferent modes of action compete against each other to a certain extent. Physicians will select the appropriate treatment based on patient characteristics”. Notably, the clinical trial for brazikumab (Allergan’s IL- 23 inhibitor pipeline) for CD aims at demonstrating its superiority versus AbbVie’s Humira, indicating that the two drugs can be used for the same groups of patients. In line with the above, respondents to the market investigation expect IL-23 inhibitors to compete with all other post-conventional treatments for UC and CD currently available on the market.42
(45) In addition, there is some fluidity across the various lines of treatments for postconventional drugs. While some EEA countries require (or encourage) the use of anti-TNFs as first-line post-conventional treatments of UC and CD for cost reasons, this is not the case in all EEA countries.
(46) In fact, the use of anti-integrins and IL-12/23 inhibitors as first-line postconventional treatments is increasing (although it is currently rather limited). This is particularly the case in UC, where Takeda’s anti-integrin Entyvio is increasingly used as a first-line post-conventional treatment.43 One KOL noted for instance that “[a]ll biologics can be prescribed as first line biologic treatment”, another one that “currently, the choice of biologic in UC or CD after conventional therapy failure is very open. Any biologics could be used. They all have advantages and limitations. Clinicians try to adapt the choice of the biologics to the profile of the patient. As there is no strong biological predictor, the clinicians rely more on the demographic and clinical characteristics of the patients”, a third that “[w]hile use of anti-TNF as first line is encouraged (biosimilar use), there is no prohibition to use of other classes in specific situations decided by most responsible physician or multidisciplinary team”.44
(47) Separately, the market investigation indicates that anti-TNFs, in addition to being used as first-line post-conventional treatments, are also largely used as second line treatment (including when a different anti-TNF was prescribed in first line), where they compete more directly with other drugs, including anti-integrins and JAK inhibitors.45
(B) Potential segmentation of the post-conventional treatments market based on the MoA
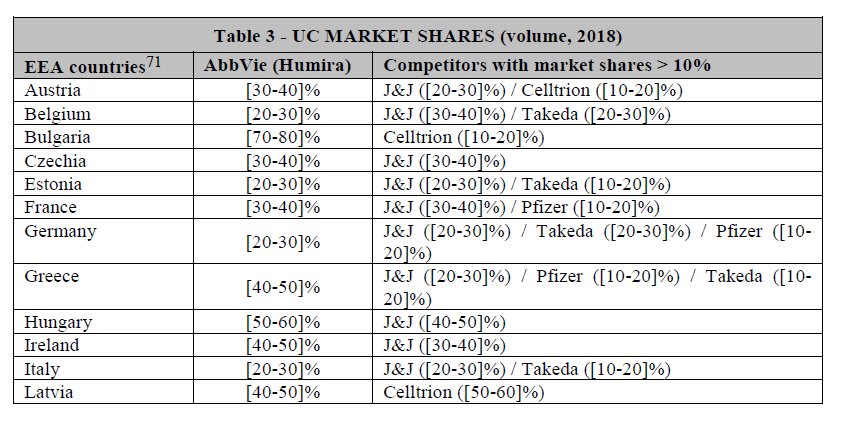
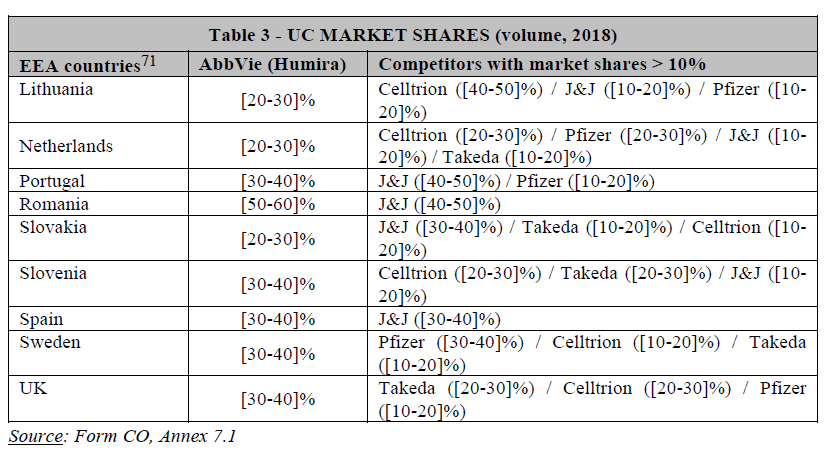
(48) Based on the results of the market investigation, it is not clear whether a segmentation based on MoA is warranted.
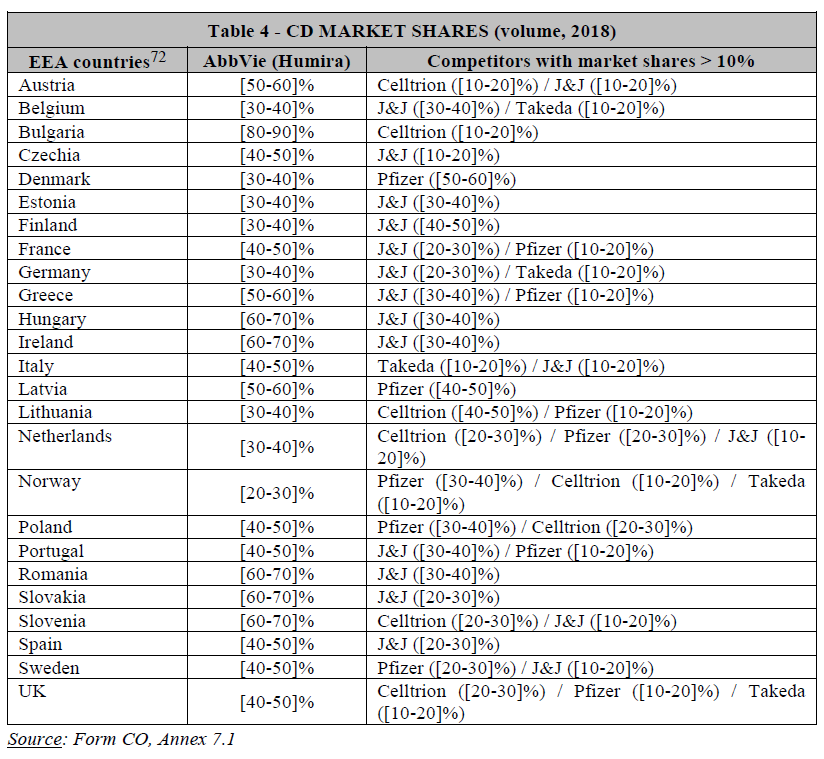
(49) On the one hand, the results of the market investigation revealed that all postconventional treatments compete with each other, regardless of their MoA (see recital 44 above).
(50) On the other hand, the Commission also found that post-conventional treatments for UC and CD based on different MoAs may not be fully substitutable. The Parties’ activities overlap in relation to IL-23 inhibitors for UC and CD, as each of AbbVie and Allergan is currently developing an IL-23 inhibitor.
(51) Drugs with different MoAs have distinct efficacy and safety profile, which are key factors for physicians when prescribing drug.46 For instance, respondents to the market investigation highlighted the promising nature of IL-23 inhibitors. Competitors rank IL-23 inhibitors consistently higher than all other available postconventional treatments, in terms of efficacy, sustainability of effects and safety, as well as in terms of speed of onset.47 Similarly, some customers and KOLs view IL- 23 inhibitors as particularly “promising” or even as products that “will change the market” due to their expected better profile.48 The distinct efficacy and safety profile of IL-23 inhibitors compared to other post-conventional drugs is further illustrated by the Parties’ internal documents […].49 Consequently, post-conventional treatments with the same MoA compete more closely with each other.50
(52) Moreover, the Commission found that, in order to be able to cover all patients’ needs, customers usually procure all different classes of post-conventional treatments available on the market (i.e. anti-TNFs, anti-integrins, IL-12/23 inhibitors and JAK inhibitors),51 which shows that drugs with different MoAs are not fully substitutable.
4.1.3.4. Conclusion
(53) Based on the above information, three alternative product market definitions giving rise to overlaps between the Parties can be envisaged, namely: (i) the market for post-conventional treatments for UC and CD, including all biologics and innovative small molecules; (ii) the market for post-conventional treatments for UC and/or CD excluding anti-TNFs; and (iii) the market for IL-23 inhibitors for UC and CD.
(54) In any event, the exact relevant product market definition for treatments of moderate to severe UC and CD can be left open. The Transaction gives rise to serious doubts as to its compatibility with the internal market regardless of whether the relevant markets are defined as: (i) all post-conventional treatments for UC and CD; (ii) all post-conventional treatments for UC and CD excluding anti-TNFs; or (iii) IL-23 inhibitors.
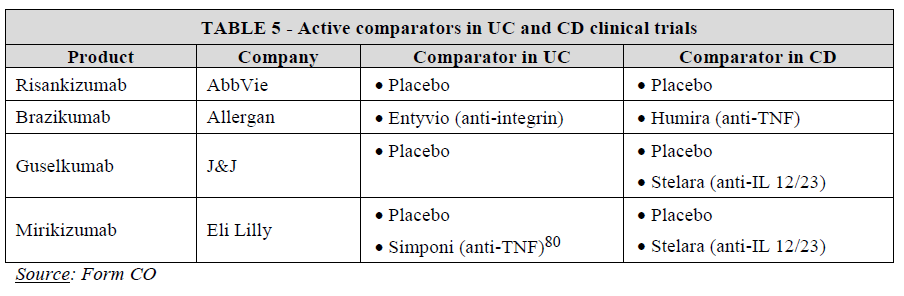
4.1.4. Competitive assessment
4.1.4.1. The Parties’ views
(55) The Parties submit that the Transaction does not give rise to competition concerns regardless of the exact scope of the market, in particular due to: (i) the crowded competitive landscape and pipeline for post-conventional treatments of UC and CD (including regarding IL-inhibitors); (ii) differences between the Parties’ timing in terms of clinical trials and market entry of pipeline products, and the uncertainty related thereto; as well as (iii) a lack of closeness of competition between the Parties’ products.
4.1.4.2. The Commission’s assessment
(A) Potential market for IL-23 inhibitors
(56) In the potential (narrowest) market for IL-23 inhibitors for the treatment of UC and CD, the Transaction gives rise to a pipeline-to-pipeline overlap between two advanced-stage pipelines of the Parties.
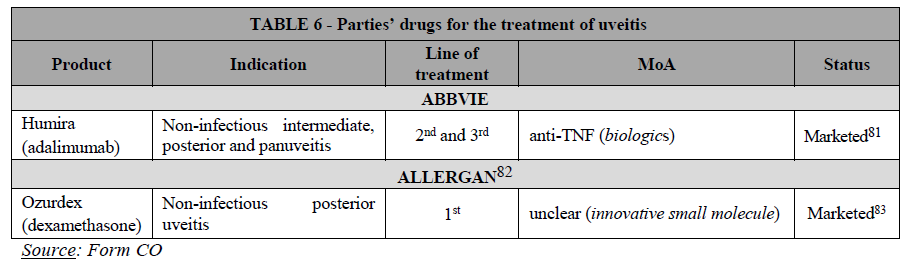
(57) As explained, AbbVie develops Skyrizi (risankizumab), currently in Phase III for UC and CD and expects to launch the drug in the EEA at the earliest after […]. Allergan develops brazikumab, currently in Phase II for UC and Phase II/III for CD and expects to launch the drug in the EEA at the earliest after […].
(58) A material number of respondents to the market investigation, and in particular the overwhelming majority of responding KOLs, raised concerns linked to the discontinuation of one of the Parties’ IL-23 inhibitors development programme (most likely brazikumab), as a result of the Transaction, the new entity having limited incentives to develop in parallel two drugs with the same MoA.52
(59) Concerns relating to the discontinuation of brazikumab by AbbVie are warranted, […].53
(60) Besides the Parties, only two other companies are currently developing IL-23 inhibitors pipelines for UC and CD, namely:
• Eli Lilly which is developing mirikizumab, currently in Phase III for UC and CD and expected to be launched in the EEA at the earliest after […]; and
• Johnson & Johnson (“J&J”), which is developing guselkumab, currently in Phase II for UC and Phase II/III for CD and expected to be launched in the EEA at the earliest after […].54
(61) As a result, the Transaction would lead to a reduction in the already limited number of players active on the market for IL-23 inhibitors from four to three, assuming that all IL-23 inhibitor pipelines reach the (EEA) market, which remains highly uncertain at this stage.55
(62) In this respect, a large number of respondents to the market investigation, including all KOLs, highlighted that it is important to have several options within the same class of post-conventional treatments (e.g. several IL-23 inhibitors).56 Reasons cited by KOLs include the possibility to obtain lower prices for the drug, supply disruption management, and better clinical data (including head-to-head comparisons), as well as a broader choice of products (including different modes of deliveries, dose intervals etc.), allowing patients to benefit from the most appropriate form of treatment at the best conditions.57
(63) As a result of potential horizontal non-coordinated effects in the plausible market for IL-23 inhibitors, in particular the potential discontinuation of Allergan’s brazikumab, leading to potentially less choice and higher prices for patients and health systems in the EEA, the Transaction raises serious doubts as to its compatibility with the internal market and the functioning of the EEA Agreement.
(B) Potential market for post-conventional treatments excluding anti-TNFs
(64) In a potential market for post-conventional treatments of UC and CD excluding anti- TNFs, the Transaction gives rise to several pipeline-to-pipeline overlaps, the main one being the overlap between the Parties’ IL-23 inhibitors.58
(65) The risk of discontinuation of brazikumab (see recitals 58-59 above) would also have a detrimental impact on this potential market for several reasons.
(66) First, the market investigation revealed the need for new alternative treatments. Most respondents confirmed the importance of having several MoA options in order to be able to cover all patients’ needs.59 There are currently only three drugs with different MoA available on this potential market, namely (i) Entyvio (vedolizumab), an antiintegrin biologic drug marketed by Takeda (for both UC and CD), (ii) Stelara (ustekinumab), an IL-12/23 inhibitor marketed by J&J (for CD and (recently) UC), and (iii) Xeljanz (tofacitinib), an innovative small molecule JAK inhibitor recently launched in the EEA by Pfizer (for UC only).
(67) According to the market investigation, these three drugs have an overall similar efficacy but different safety profiles.60 Anti-integrins (Entyvio) are considered to be the safest drug currently available on the market (i.e. it leads to less adverse effects and a lower risk of infection), due to a more targeted (gut-specific) MoA. IL-12/23 inhibitors (Stelara) are associated with a risk of immunosuppression. JAK inhibitors (Xeljanz) are characterized by a particularly poor safety profile61 and safety warnings.62
(68) In addition, there are a number of post-conventional treatments for UC and/or CD currently under development but, as illustrated in the table below, most of them relate to existing MoAs.
(69) Second, IL-23 inhibitors are expected to have a better efficacy and safety profile than (marketed and pipeline) drugs with other MoAs. In particular, KOLs and competitors expect IL-23 inhibitors to have a better safety profile than existing postconventional treatments, including anti-integrins, which is a key consideration for prescribing practitioners. For instance, some customers and KOLs view IL-23 inhibitors as particularly “promising” or even as products that “will change the market” due to their expected better profile.63 This is also corroborated by the Parties’ internal documents, […].64 Therefore, within the potential market for postconventional treatments for UC and/or CD excluding anti-TNFs, the Parties’ IL-23 inhibitors are expected to be close competitors.65
(70) Third, the discontinuation of Allergan’s brazikumab would remove a promising competitive constraint on the market for UC and CD post-conventional treatments excluding anti-TNFs, compared to the situation absent the Transaction.
(71) Indeed, Allergan implemented a strategy to differentiate brazikumab from competing products, including in particular head-to-head trials comparing the efficacy and safety of its IL-23 inhibitor pipeline drug with Takeda’s anti-integrin (Entyvio) in UC, i.e. the leading post-conventional treatment for UC and CD excluding anti- TNFs. Allergan’s head-to-head trial against Entyvio aims at establishing a clinical superiority of brazikumab over Entyvio, as evidenced by Allergan’s internal documents.66 Moreover, Allergan is also testing brazikumab with an IL-22 companion diagnostics in order to assess whether the compound is more effective in patients expressing higher levels of the IL-22 biomarker.
(72) The Commission found that, if successful, brazikumab’s head-to-head trials against Entyvio in UC and Humira in CD and IL-22 biomarker indication would give Allergan a competitive advantage compared to competitors. Discontinuing such head-to-head trials and biomarker indication would potentially deprive practitioners from useful clinical data, ultimately resulting in a lower quality of patient treatment. For instance, one KOL noted that “the head-to-head comparisons may be used as one of the strongest arguments (if not the strongest) for positioning of brazikumab” and that “if the use of IL22 as a companion diagnostic holds true it would probably be rapidly picked up and implemented at IBD-specialised centres […]”; another one that “[i]t is imperative to have head-to-head comparisons in order to identify the efficacy, safety, sustainability long-term of a new agent and position this new drug in the therapeutic algorithms of IBD”.67
(73) As a result of potential horizontal non-coordinated effects in the plausible market for post-conventional treatments for UC and CD excluding anti-TNFs in the EEA, in particular the potential discontinuation of Allergan’s IL-23 inhibitor, leading to potentially less choice, lower quality treatments, and higher prices for patients and health systems in the EEA, the Transaction raises serious doubts as to its compatibility with the internal market and the functioning of the EEA Agreement.
(C) Potential market including all post-conventional treatments
(74) In a potential market for all post-conventional treatments of UC and CD, the Transaction gives rise to several market-to-pipeline68 and pipeline-to-pipeline overlaps,69 the most relevant ones being (i) the market-to-pipeline overlap between AbbVie’s Humira (anti-TNF) and Allergan’s brazikumab (IL-23 inhibitor); as well as (ii) the pipeline-to-pipeline overlap between the Parties’ IL-23 inhibitors.
(75) On this potential market, the Transaction also raises serious doubts as to the compatibility of the Transaction with the internal market and the functioning of the EEA Agreement for several reasons.
(76) First, the market investigation revealed Humira’s leadership on the market of postconventional treatments.
(77) Anti-TNFs are the first generation of biologics used in UC/CD. There are a number of marketed anti-TNFs in the EEA, besides AbbVie’s Humira (adalimumab), which are indicated for the treatment of UC and CD, including J&J’s Remicade (infliximab) and Simponi (golimumab), as well as adalimumab and infliximab biosimilars following Humira’s loss of exclusivity in 2018,70 and Remicade’s loss of exclusivity between 2013 and 2015. Simponi is scheduled to lose exclusivity in 2024.
(78) AbbVie currently markets its anti-TNF Humira, the first post-conventional treatment for UC and CD, in […].
(79) Table 3 below provides an estimate of AbbVie’s market share in the EEA member states for the treatment of moderate to severe UC, based on the number of patients treated, where such share exceeds 20%.
(80) Table 4 below provides an estimate of AbbVie’s market share in the EEA member states for the treatment of moderate to severe CD, based on the number of patients treated, where such share exceeds 20%.
(81) Humira is a blockbuster drug ([…] sales in the EEA in 2018), which accounts for over 60% of AbbVie’s worldwide revenues. Since the loss of its exclusivity in 2018 and the launch of several biosimilars, Humira’s market shares have generally declined in Europe, […]. Humira’s market shares are expected to decrease further over the next few years, due to the health authorities’ incentives to increase the use of biosimilars, priced lower than Humira.
(82) However, AbbVie still retains a significant market presence in numerous EEA countries (see Tables 3 and 4 above). In particular, its market share exceeds 50% in Austria (in CD), Bulgaria (in UC and CD), Greece (in CD), Hungary (in UC and CD), Ireland (in CD), Latvia (in CD), Romania (in UC and CD), Slovakia (in CD) and Slovenia (in CD) in 2018. AbbVie’s market share has even increased in a number of EEA countries between 2018 and 2019, in spite of biosimilar entry in the EEA, including in [seven EEA countries in UC and four EEA countries in CD].73 According to well-established case law, such large market shares may in themselves be evidence of the existence of a dominant position.74
(83) The market investigation also revealed that Humira benefits from a long-lasting and strong track-record in the treatment of UC and CD. Such track-record is an important criterion for physicians prescribing post-conventional treatments to patients.75 According to a large majority of customers, AbbVie’s track-record in selling Humira provides the company with a strong competitive advantage since competing products do not benefit from a similar track record. This advantage is linked in particular to the company’s long-term experience with biologics in the IBD field, established customer base, contacts with physicians, KOLs and public health authorities.76
(84) Several competitors also share this view, with one stating for instance that “AbbVie has a long track record in the disease area and strong commercial presence. It has the #1 selling biologic across all indications and also specifically in IBD”.77 Another competitor also indicated that “Humira has solid long-term safety data as well as real-world evidence, compared to the other post-conventional treatments for UC and CD (which have been on the market for a shorter period of time)”.78
(85) Second, the number of competitors active at national level is limited in several EEA countries (e.g. duopoly or quasi duopoly in Hungary, Ireland and Romania) (see Tables 3 and 4 above).
(86) Third, as previously mentioned, the market investigation also confirmed the closeness of competition between the Parties’ IL-23 inhibitors and the risk of discontinuation of brazikumab. The Transaction may therefore lead to the elimination of a potentially important competitive constraint.
(87) Indeed, the market investigation confirmed that IL-23 inhibitors are a promising class, especially brazikumab, which is currently undergoing several head-to-head trials against existing treatments, namely Entyvio in UC and Humira in CD with a view of proving superiority of brazikumab over these products, as evidenced by Allergan’s internal documents.79 As illustrated in Table 5 below, only Allergan conducts head-to-head trials to show superiority of brazikumab with the current standard of care in UC (Takeda’s Entyvio) and CD (AbbVie’s Humira). If successful, these trials would constitute a competitive advantage for brazikumab.
(88) As a result of potential horizontal non-coordinated effects in the plausible market for post-conventional treatments for UC and CD in the EEA (in particular (i) the strengthening of AbbVie’s dominant position; and (ii) the potential discontinuation of Allergan’s IL-23 inhibitor, both leading to potentially less choice and higher prices for patients and health systems), the Transaction raises serious doubts as to its compatibility with the internal market and the functioning of the EEA Agreement.
4.1.4.3. Conclusion
(89) It derives from the above that the Transaction raises serious doubts as to its compatibility with the internal market and the functioning of the EEA Agreement as a result of the overlap of the Parties’ activities in post-conventional treatments for UC and CD, under all plausible market definitions.
4.2. Treatments for Uveitis
(90) Uveitis is a generic term covering inflammation of the interior part of the eye (as opposed to eye surface inflammations such as conjunctivitis), also referred to as uvea. Uveitis can cause eye pain, eye redness, sensitivity to light and can lead to blindness.
(91) Uveitis can be classified as anterior, intermediate, or posterior depending on the part of the uvea affected by the inflammation (or as panuveitis when the entire uvea is affected). Uveitis may be acute or chronic, and may be infectious or not.
(92) Different treatments are available depending on the type (infectious or noninfectious) and location of the uveitis (anterior, intermediate, posterior uveitis, or panuveitis).
4.2.1. The Parties’ activities
(93) The Parties’ drugs for the treatment of uveitis are detailed in Table 6 below.
(94) Allergan markets one product approved for the treatment of uveitis in the EEA, Ozurdex, a corticosteroid that is indicated for the treatment of posterior noninfectious uveitis. […].
(95) AbbVie has one marketed product, its anti-TNF drug Humira, indicated for the treatment of posterior non-infectious uveitis only when a corticosteroid treatment has had an inadequate response (or is inappropriate). […].
4.2.2. Market definition
(96) As regards product market definition, the Commission has not assessed the market for the treatment of uveitis in the past. In line with the Parties’ views, the Commission considers that, for the purpose of this Decision, the exact scope of the market for uveitis treatments can be left open as the Transaction does not give rise to serious doubts as to its compatibility with the internal market even under the narrowest market definition giving rise to an overlap between the Parties’ drugs (i.e. market for the treatments of non-infectious posterior uveitis).
(97) As regards the geographic market definition, the Commission has consistently considered the market for marketed drugs to be national in scope.84 The Parties do not contest this. Nothing in the market investigation suggests that the Commission should depart in the present case from its previous practice.
4.2.3. Competitive assessment
(98) The Parties argue that no competition concerns arise in the market for uveitis treatments or any of its plausible sub-segmentations, in particular because: (i) the products are not prescribed to patients as alternative treatments; (ii) the products have different ATC3 codes; (iii) the Parties compete more closely with other alternative treatments for uveitis.
(99) The Commission considers that the Transaction does not raise serious doubts as to its compatibility with the internal market and the functioning of the EEA agreement.
(100) First, the Parties’ products for the treatment of uveitis are very differentiated. They (i) rely on different active ingredient, (ii) involve different MoAs (see Table 6 above), (iii) are used at different stages of a patient’s treatment (corticosteroids, such as Allergan’s Ozurdex, are used as first line treatments whereas AbbVie’s Humira is generally used as second or third line treatment), and (iv) have different MoD (Allergan’s Ozurdex is administered directly in the eye85 whereas AbbVie’s Humira is injected subcutaneously). The products further differ in terms of potential side effects (local for Ozurdex, against immunosuppression for Humira) and treatment duration, as corticosteroids such as Ozurdex typically cannot be prescribed over a prolonged period of time. As a result, the Parties’ products are not close competitors in this space.
(101) The market investigation confirmed that steroids such as Ozurdex and biologics such as Humira are unlikely to be substitutes for the treatment of uveitis. In particular, one KOL indicated that “Ozurdex and Humira are not alternatives to each other. These two products are used in a sequential fashion, in different cases, and are not substitutable”.86
(102) Second, the Parties will continue to face the competition of many alternative products, including from drugs competing more closely with each of the Parties’ products. There are various competing corticosteroids besides Ozurdex, including Iluvien, which is indicated for uveitis and has a longer durability compared to Ozurdex.87 While Humira is the only anti-TNF approved for the treatment of uveitis, there are biosimilars drugs available on the market. Furthermore, other anti-TNFs may be used off-labels. This is corroborated by the fact that […].
(103) Finally, the results of the market investigation did not reveal any substantiated concerns as regards the overall impact of the Transaction in the markets for uveitis treatments in the EEA.
4.2.4. Conclusion
(104) For all these reasons, the Commission concludes that the Transaction does not give rise to serious doubts as to its compatibility with the internal market and the functioning of the EEA Agreement as regards its impact on competition in the possible markets for uveitis treatments and its plausible sub-segmentations.
5. COMMITMENTS
5.1. Framework for the assessment of the Commitments
(105) Where a concentration raises serious doubts as regards its compatibility with the internal market, the parties may undertake to modify the concentration to remove the grounds for the serious doubts identified by the Commission. Pursuant to Article 6(2) of the Merger Regulation, where the Commission finds that, following modification by the undertakings concerned, a notified concentration no longer raises serious doubts, it shall declare the concentration compatible with the internal market pursuant to Article 6(1)(b) of the Merger Regulation.
(106) As set out in the Commission’s Remedies Notice,88 the commitments have to eliminate the competition concerns entirely, and have to be comprehensive and effective from all points of view.89
(107) In assessing whether commitments will maintain effective competition, the Commission considers all relevant factors, including the type, scale and scope of the proposed commitments, with reference to the structure and particular characteristics of the market in which the transaction is likely to significantly impede effective competition, including the position of the parties and other participants on the market.90
(108) In order for the commitments to comply with those principles, they must be capable of being implemented effectively within a short period of time. Concerning the form of acceptable commitments, the Merger Regulation gives discretion to the Commission as long as the commitments meet the requisite standard. Structural commitments will meet the conditions set out above only in so far as the Commission is able to conclude with the requisite degree of certainty, at the time of its decision, that it will be possible to implement them, and that it will be likely that the new commercial structures resulting from them will be sufficiently workable and lasting to ensure that the serious doubts are removed.91 Divestiture commitments are normally the best way to eliminate competition concerns resulting from horizontal overlaps.
5.2. Proposed Commitments
(109) In order to render the concentration compatible with the internal market, the Parties submitted a set of commitments under Article 6(2) of the Merger Regulation on 10 December 2019 (the “Commitments”).
(110) The Commission market tested the Commitments to assess whether they are sufficient and suitable to remedy the serious doubts identified in Section 4 of this Decision. The feedback received during the market test confirmed that the Commitments eliminate the Commission’s competition concerns entirely.
(111) The Commitments are annexed to this Decision and form an integral part thereof.
5.2.1. Description of the Commitments
(112) In order to dispel the serious doubts raised by the Commission, the Parties submitted commitments consisting of a full divestiture of the development, manufacturing and marketing rights related to Allergan’s IL-23 inhibitor pipeline programme (brazikumab) at worldwide level (the “Divestment Business”) to a suitable purchaser (the “Purchaser”).
(113) Brazikumab is being developed by Allergan pursuant to a licence agreement with AstraZeneca under which AstraZeneca granted Allergan an exclusive licence to develop and commercialise brazikumab worldwide. Brazikumab is currently in Phase II clinical trials for the treatment of UC and Phase II/III clinical trials for the treatment of CD. Brazikumab’s clinical trials include head-to-head trials to assess whether brazikumab is more effective than existing approved treatments (namely AbbVie’s Humira in CD and Takeda’s Entyvio in UC). Allergan is also testing brazikumab with an IL-22 companion diagnostics in order to assess whether the compound is more effective in patients expressing higher levels of the IL-22 biomarker. Furthermore, Allergan is considering […] in relation to brazikumab.
(114) The Divestment Business includes assets necessary to conduct and complete brazikumab’s global clinical trials, obtain the required marketing authorisation from regulatory authorities (if the trials are successful) and bring the product to the EEA and (potentially) non-EEA markets. In particular, the Divestment Business includes the following main intangible and tangible assets: (i) rights to conduct the clinical trials, to manufacture and to market brazikumab globally; (ii) patents, copyrights, data and know-how related to brazikumab; (iii) the proposed “brazikumab” international non-proprietary name; (iv) licences, permits and authorisations issued by third parties in relation to brazikumab; (v) the relevant reports related to the clinical trials, regulatory files, books and records, and the documentation related to marketing plans and forecasts of brazikumab; (vi) contracts with third parties related to brazikumab, to the extent these are transferrable; (vii) all inventories of brazikumab; and (vii) certain Allergan employees working on the brazikumab programme.
(115) Furthermore, with the aim of ensuring a smooth transfer of the Divestment Business, the Parties committed to offer a number of transitional supply arrangements to the Purchaser, including: (i) the supply of any transitional support required by the Purchaser to ensure the continuity of the clinical trials (e.g. technical assistance, liaising with regulatory authorities), for up to […]; (ii) the supply of all products and services currently provided by Allergan to the Divestment Business, for up to […]; and (iii) the supply of products and services provided to Allergan by third parties, through back-to-back arrangements, if the contracts in question cannot be transferred, for up to […].
(116) In addition to the standard purchaser requirements contained in the Commission’s template for divestiture remedies, the Commitments provide that the Purchaser must have: (i) the incentive to pursue the clinical trials for the EEA approval of brazikumab as they are currently designed; (ii) expertise and experience in the clinical development of medicinal products for EEA approval; (iii) expertise and experience in having interactions with relevant EEA-wide and national bodies that decide on approval of medicinal products in the EEA; and (iv) experience in the pricing and reimbursement, marketing, promotion, sales and distribution of medicinal products in the EEA, or the ability to develop such capabilities for the marketing of brazikumab.
(117) The Commitments also provide that the Parties should enter into commitments related to the divestiture, inter alia regarding the separation of the Divestment Business from the Parties’ retained businesses, the preservation of the viability, marketability and competitiveness of the Divestment Business, as well as the appointment of a monitoring trustee and, if necessary, a divestiture trustee.
(118) Finally, the Commitments provide for an upfront buyer provision pursuant to which the Transaction cannot be implemented until the Commission has given its approval to the purchase of the Divestment Business by the Purchaser.
(119) The Parties argued that the Commitments remedy the Commission’s competition concerns since the pipeline-to-pipeline overlap between the Parties’ IL-23 inhibitor pipeline drugs will be removed entirely. The Parties further argued that the rights and assets to be transferred are sufficient for a Purchaser to continue the development and, depending on the outcome of the clinical trials, ultimately market brazikumab, thus allowing the Purchaser to operate the Divestment Business as a viable and independent business in the EEA. In addition, the Parties explained that the earliest date currently anticipated by Allergan for the launch of brazikumab in the EEA […], which gives the Purchaser […] to prepare and establish the necessary operations needed for the commercialisation of brazikumab.92
5.2.2. Results of the market test
(120) The market test was launched on 11 December 2019 and sought to assess the scope and effectiveness of the Commitments, the viability and attractiveness of the Divestment Business, and the appropriateness of the purchaser suitability criteria.
(121) The majority of competitors that responded to the market test considered the Divestment Business to be an attractive asset, likely to appeal to suitable purchasers. In fact, several of the respondents expressed interests in acquiring brazikumab.93
(122) In terms of scope, market participants considered that the Divestment Business includes all the necessary assets, for a purchaser to be able to continue the clinical trials and ultimately bring the product to the EEA market.94 More specifically, market participants confirmed that the personnel transferred was sufficient to continue the development of the programme and that the Key Personnel as identified in the Commitments covered all the necessary roles.95
(123) As concerns the Purchaser, the results of the market test confirmed the relevance of the purchaser requirements included in the Commitments. In particular, most market participants indicated that the most suitable type of purchaser would be a company with (i) expertise and experience in dealing with national bodies in the EEA that decide on pricing and reimbursement, and (ii) capabilities in the commercialisation and distribution of pharmaceutical products in the EEA.96 However, a majority of competitors also stated that, in the present case, a pharmaceutical company could rely on third parties (including contract research organisations) based in the EEA to develop a pipeline product such as the Divestment Business.97 Furthermore, the vast majority of market participants consider that brazikumab’s head-to-head trials and IL-22 biomarker indication are particularly important,98 which confirms the need to pursue the clinical trials as they are currently designed.
(124) As regards potential implementation risks, market participants generally consider that the transfer of the Divestment Business and, in particular, the transfer of thirdparty supply contracts does not entail particular hurdles.99
(125) Finally, the respondents who expressed an opinion generally consider that the proposed transitional agreements provided for in the Commitments were sufficient, notably in terms of duration, to ensure a smooth transfer of the Divestment Business.100
5.2.3. Commission’s assessment
(126) The Commitments remove the entire pipeline-to-pipeline and market-to-pipeline overlaps between the Parties’ activities for which the Commission raised serious doubts following the results of the Phase I market investigation. In particular, the Commitments include the tangible and intangible assets necessary to conduct and complete the clinical trials at global level for brazikumab, and, if successful, obtain a marketing authorisation and bring brazikumab to the market.
(127) The Commitments also include potential transitional agreements, which are sufficient, both in terms of scope and duration, to ensure a smooth transfer of the Divestment Business to the Purchaser. Furthermore, the Commitments include targeted purchaser criteria, such that the Divestment Business will be transferred to a suitable purchaser. Finally, the Commission considers that the upfront buyer clause mitigates implementation risks inherent to the development of pipeline drugs.
(128) The results of the market test confirmed the suitability and viability of the Divestment Business and did not reveal any shortcomings of the Commitments.
(129) On this basis, and in view of the presence of a number of interested potential purchasers, the Commission considers that the Divestment Business is attractive and likely to be acquired by a suitable purchaser.
(130) For the reasons outlined above, the Commission concludes that the Commitments are sufficient in scope and suitable to eliminate the serious doubts as to the compatibility of the Transaction with the internal market on all plausible markets.
6. CONDITIONS AND OBLIGATION
(131) Under the first sentence of the second subparagraph of Article 6(2) of the Merger Regulation, the Commission may attach to its decision conditions and obligations intended to ensure that the undertakings concerned comply with the commitments they have entered into vis-à-vis the Commission with a view to rendering a notified concentration compatible with the internal market.
(132) The achievement of the measure that gives rise to the structural change of the market is a condition, whereas the implementing steps which are necessary to achieve this result are generally obligations on the Parties. Where a condition is not fulfilled, the Commission’s decision declaring the concentration compatible with the internal market no longer stands. Where the undertakings concerned commit a breach of an obligation, the Commission may revoke the clearance decision in accordance with Article 8(6) of the Merger Regulation. The undertakings concerned may also be subject to fines and periodic penalty payments under Articles 14(2) and 15(1) of the Merger Regulation.
(133) In accordance with the distinction described above, the Decision in this case is conditioned on the full compliance with the requirements set out in Section B of the Commitments (including the Schedule), which constitute conditions. The remaining requirements set out in the other section of the Commitments constitute obligations on the Parties.
(134) The detailed text of the Commitments is annexed to this Decision. The full text of the Commitments forms an integral part of this Decision.
7. CONCLUSION
(135) For the above reasons, the Commission has decided not to oppose the notified operation as modified by the commitments and to declare it compatible with the internal market and with the functioning of the EEA Agreement, subject to full compliance with the conditions in Section B of the Commitments annexed to the present decision and with the obligations contained in the other sections of the said Commitments. This decision is adopted in application of Article 6(1)(b) in conjunction with Article 6(2) of the Merger Regulation and Article 57 of the EEA Agreement.
FOOTNOTES :
1 OJ L 24, 29.1.2004, p. 1 (the “Merger Regulation”). With effect from 1 December 2009, the Treaty on the Functioning of the European Union (“TFEU”) has introduced certain changes, such as the replacement of “Community” by “Union” and “common market” by “internal market”. The terminology of the TFEU will be used throughout this decision.
2 OJ L 1, 3.1.1994, p. 3 (the “EEA Agreement”).
3 Turnover calculated in accordance with Article 5(1) of the Merger Regulation and the Commission Consolidated Jurisdictional Notice (OJ C95, 16.4.2008, p. 1).
4 FDPs refer to the finished dosage form of pharmaceutical products, which, in other words, are ready to be used by customers. FDPs contain (i) an active pharmaceutical ingredient (or “API”, which correspond to the component present within the product that provides the pharmacological action in the body, e.g. acetyl salicylic acid in an aspirin tablet), or a combination of APIs and (ii) other excipients.
5 See, for example, cases M.8974 – Procter & Gamble / Merck Consumer Health Business, decision of 27.8.2018; M.7919 – Sanofi/Boehringer Ingelheim Consumer healthcare Business, decision of 4.8.2016; M.6969 – Valeant Pharmaceuticals International/Bausch & Lomb Holdings, decision of 5.8.2013; M.5778 – Novartis/Alcon, decision of 9.8.2010; M.7276 – GlaxoSmithKline/ Novartis Vaccines Business (Excl. Influenza) / Novartis Consumer Health Business, decision of 28.1.2015; M.5865 – Teva / Ratiopharm, decision of 3.8.2010; and M.8889 – Teva / PGT OTC, decision of 29.1.2010.
6 OJ C 372, 9.12.1997, p. 5.
7 See case M.9274 – GSK/Pfizer Consumer Healthcare Business, decision of 10.7.2019, recital 15 and case M.7275 - Novartis/GSK Oncology, decision of 28.1.2015, recitals 207 and 216.
8 See case M.5476 - Pfizer/Wyeth, decision of 17.7.2009, recitals 21-26 (renal cell carcinoma).
9 See case M.8955 - Takeda/Shire, decision of 20.11.2018, recitals 9-49 (inflammatory bowel diseases).
10 See case M.7275 - Novartis/GSK Oncology, decision of 28.11.2015, recital 31. The Commission found that, given the different ways in which they treat cancer, chemotherapies, targeted therapies, and immunotherapies may not be substitutable and they are most often used as complementary treatments. For instance, chemotherapies and targeted therapies attack cancerous cells directly, whereas immunotherapies work to enable the patient’s own immune system to attack the cancer. Targeted therapies are designed to interact with a specific target associated with cancer, whereas chemotherapies are typically identified because they kill rapidly dividing cells. Targeted therapies and immunotherapies are also typically more expensive than chemotherapies. Given their novelty, targeted therapies and immunotherapies are generally still under patent, while many chemotherapy drugs are off patent.
11 See case M.8955 - Takeda/Shire, decision of 20.11.2018, recitals 17-25.
12 See case M.7275 - Novartis/GSK Oncology, decision of 28.1.2015, recitals 33 and 143. Line of treatment refers to the setting for which a specific drug is indicated. For example, a drug indicated for second-line of treatment should be used only after another therapy (the first-line of treatment) has proven ineffective or if this other therapy cannot be prescribed altogether to a specific patient.
13 See case M.8955 - Takeda/Shire, decision of 20.11.2018, recitals 31-49.
14 See case M.8401 - J&J/Actelion, decision of 9.06.2017, recital 60.
15 See cases M.3354 - Sanofi-Synthelabo/Aventis, decision of 26.4.2004, recital 57; and M.7275 - Novartis/GSK Oncology, decision of 28.1.2015, recitals 85-94. ; 16 Case M.7275 - Novartis/GSK Oncology, decision of 28.1.2015, recital 26.
17 See cases M.8955 - Takeda/Shire, decision of 20.11.2018, recital 56 and M.8401 - J&J/Actelion, decision of 9.06.2017, recital 66.
18 See most recently, case M.8955 - Takeda/Shire, decision of 20.11.2018, recital 56.
19 See case M.8084 - Bayer/Monsanto, decision of 21.3.2018, recital 48. The remainder of this decision focuses on horizontal, non-coordinated effects, as the Transaction does not give rise to non-horizontal overlaps or coordinated effects. Regarding the framework of assessment of horizontal non-coordinated effects, see the Commission Guidelines on the assessment of horizontal mergers under the Merger Regulation (the “Horizontal Merger Guidelines”) OJ C 31, 5.2.2004, p. 5, paragraphs 24-38.
20 Horizontal Merger Guidelines, paragraph 8.
21 Horizontal Merger Guidelines, paragraph 38.
22 In the pharmaceutical industry, pipeline drugs go through several development stages, starting with preclinical trials in laboratories and on animals, and later moving on to clinical trials in humans (so called “Phase I”, “Phase II” and “Phase III” clinical trials). Clinical trials in humans are strictly regulated in order to ensure the protection of trial subjects and the credibility of the results. In most jurisdictions, before a clinical trial can start, the sponsor must typically apply for and receive clinical trial authorisation from the competent authorities. The clinical trial is also typically registered on public databases. All clinical trial protocols conducted in Europe are identified at European level with a unique number and registered in the European Clinical Trials Database (EudraCT database). ClinicalTrials.gov is a webbased resource that provides easy access to information on publicly and privately supported clinical studies conducted worldwide. Unlike clinical trials, preclinical trials are not registered in public databases and their existence is not always disclosed to the public.
23 See case M.7275 - Novartis/GSK Oncology, decision of 28.1.2015, recital 90 and M.9294 – BMS/Celgene, decision of 29.7.2019, recital 21.
24 See case M.7275 - Novartis/GSK Oncology, decision of 28.1.2015, recitals 89-90 and M.9294 – BMS/Celgene, decision of 29.7.2019, recital 22.
25 See cases M.7932 - Dow/Dupont, decision of 27.3.2017, recitals 272-302 and M.8084 - Bayer/Monsanto, decision of 21.3.2018, recitals 48-54.
26 The phases of clinical development for pipeline products can be described as follows. Phase I starts with the initial administration of a new drug into humans, with trials carried out on a small number of people (e.g. in oncology, the sample size is usually in the low tens). The focus of Phase I trials is to confirm that the drug is safe to use in humans and to identify the appropriate dosage and exposure-response relationship. They typically involve one or a combination of the following aspects: estimation of initial safety and tolerability, characterisation of a drug's absorption, distribution, metabolism, and excretion, and early measurement of drug activity. Phase II usually starts with the initiation of studies to explore therapeutic efficacy in patients. Studies in Phase II are typically conducted on a small group of patients (generally around 20 to 50 up to some hundreds per cohort or treatment arm) that are selected based on stricter criteria for indications. Phase III trials aim to demonstrate or confirm therapeutic benefit in a larger group of patients (Phase III trials will typically have hundreds of patients and may have over a thousand, for example for autoimmune diseases). Studies in Phase III are designed to confirm the preliminary evidence accumulated in Phase II that a drug is safe and effective for use in the intended indication and recipient population. Usually, Phase III trials will involve a comparison of the investigational agent with a placebo or the standard of care therapy. These studies are also intended to provide an adequate basis for marketing approval. Phase IV begins after drug approval to monitor possible adverse reactions and/or new side effects over time. See Case M.8401 - J&J/Actelion, decision of 9.06.2017, footnote 6 and M.9294 – BMS/Celgene, decision of 29.7.2019, footnote 27.
27 The Transaction does not raise competition concerns in this respect given the very large number of R&D organisations competing at global level (e.g., pharmaceutical and biotechnology companies, university research programmes) in autoimmune diseases, where the Parties’ activities mainly overlap. A report of the Pharmaceutical Research and Manufacturers of America (“PhRMA”) indicated that in 2016, there were 311 medicines and vaccines in development by more than 150 companies for patients with autoimmune diseases (see http://phrma-docs.phrma.org/sites/default/files/pdf/medicines-in-developmentreport- autoimmune-diseases.pdf).
28 The Transaction also gives rise to potential minor horizontal overlaps in relation to (i) Parkinson’s disease, (ii) Alzheimer’s disease, (iii) cystic fibrosis and (iv) oncology […], where it is unlikely to raise serious doubts as to its compatibility with the internal market for the following reasons.
In cystic fibrotic and oncology, the overlaps arising from the Transaction only relate to marketed or pipeline products for which Allergan has no development, marketing and distribution rights […]. Moreover, the Parties’ products are differentiated (with different MoAs, therapeutic uses and targeted patient populations). Furthermore, the Parties face several competing products for the same indications. Also, some of the Parties’ drugs are pipelines in early stages of development (Phase I), i.e. many years away from a hypothetical launch on the market, which remains highly uncertain.
In Alzheimer’s disease, AbbVie has one pipeline product (in Phase II) and options on two other pipelines (in Phase I); Allergan has two pipeline products (in […]). The Parties’ drugs are complementary rather than substitutes since AbbVie’s products are […] treatments, while Allergan’s products are […]. Also, the Parties’ products have different MoAs, face a large number of competitors. Moreover, Allergan’s product and the two products on which AbbVie has an option are pipeline drugs in early stages of development.
In Parkinson’s disease, AbbVie has one marketed product in the EEA (Duopdopa) and two pipeline drugs (in Phase I and III). […], in some limited instances, its Botox drug can be used to treat a number of symptoms associated with this disease (e.g. movement problems, drooling and incontinence), However,
Botox does not act on cerebral pathways and, therefore, does not constitute a substitute for Parkinson’s disease treatments but a complement, which is only prescribed in conjunction with other therapies.
29 See cases M.7339 – AbbVie/Shire, decision of 16.10.2014 (this transaction ultimately did not materialise) and M.8955 – Takeda/Shire, decision of 20.11.2018.
30 See cases M.7339 – AbbVie/Shire, decision of 16.10.2014, recital 27 and M.8955 – Takeda/Shire, decision of 20.11.2018, recitals 25 and 30.
31 See case M.8955 – Takeda/Shire, decision of 20.11.2018, recitals 31-49.
32 See case M.9294 – BMS/Celgene, decision of 29.7.2019, recital 120.
33 Questionnaire Q1 to customers, replies to questions 16 and 28. Questionnaire Q2 to competitors, replies to questions 4.2, 5.2, 6.2, 10.1, 12.1, 14.1, 15.1, 19.1, 22.1.1, 22.2.1, 23.1, 25.1, 26.1, 27.1.1, 27.1.1, 27.1.1, 27.1.1, 27.1.1, 27.1.1, 27.1.1, 28.1, 31.1, 34.2 and 35.2.
34 Questionnaire Q1 to customers, replies to question 7. Questionnaire Q2 to competitors, replies to question 9. Questionnaire to Key Opinion Leaders (“KOLs”), replies to question 3(a).
35 As a result, Allergan’s Asacol is not substitutable with AbbVie’s Humira or any other post-conventional treatments. This is confirmed by all KOLs who responded to the market investigation. See Questionnaire to KOLs, replies to question 3(b). Therefore, Allergan’s Asacol does not overlap with AbbVie’s treatments for UC and CD and will not be discussed further in the Decision.
36 Questionnaire Q2 to competitors, replies to questions 7.1 and 7.2.
37 Questionnaire Q2 to competitors, replies to question 8. Questionnaire to KOLs, replies to question 6.
38 Questionnaire to KOLs, replies to question 2.
39 Questionnaire Q1 to customers, replies to questions 27.2, 27.3, and 27.4.
40 Questionnaire Q1 to customers, replies to question 27.1. In this respect, one competitor also explained that “IL-23 products are expected to compete initially with Stelara (Il-12/23), and other mechanism of actions such as Entyvio, Xeljanz, Upadacitinib and Ozanimod as 2nd or 3rd initially. Later, with the physician gaining clinical experience with IL-23, these products could be expected to compete as 1st line therapy with anti-TNFs”, questionnaire Q2 to competitors, reply to question 18.
41 Questionnaire Q2 to competitors, replies to question 25. Questionnaire to KOLs, replies to question 4.
42 Questionnaire Q2 to competitors, replies to question 18.
43 Questionnaire Q1 to customers, replies to question 27.2. For instance, one customer expects “antiintegrin market share to increase slightly or stagnate overall, there will be increased use as first line but decreased in the second and third line space”. Questionnaire Q2 to competitors, replies to question 26. See non-confidential minutes of call with KOL dated 18 September 2019.
44 Questionnaire Q2 to competitors, replies to question 8. Questionnaire to KOLs, replies to question 6.
45 Questionnaire Q2 to competitors, replies to questions 7.1 and 7.2.
46 Questionnaire Q2 to competitors, replies to question 6.
47 Questionnaire Q2 to competitors, replies to questions 27.1 to 27.8.
48 Questionnaire Q1 to customers, replies to question 15. Non-confidential minutes of call with KOL dated 16 September 2019, para. 20. Non-confidential minutes of call with KOL dated 17 September 2019, para. 15. Non-confidential minutes of call with KOL dated 18 September 2019, para. 18: “IL-23 inhibitors are very promising and have the potential to be a game-changer for the treatment of IBD, with a high level of patients achieving clinical remission (40-50%)”.
49 See for instance Annex 7.4 to Form CO.
50 Conversely, respondents to the market investigation generally do not expect a significant level of differentiation among IL-23 inhibitors, although noting that the products are not yet marketed and that additional studies may highlight differentiating features. As a result, no sub-segmentation of the potential market for IL-23 inhibitors will be considered in the present Decision. See Questionnaire to KOLs, replies to question 8.
51 Questionnaire Q1 to customers, replies to question 20.
52 Questionnaire Q1 to customers, replies to questions 12 and 31. Questionnaire Q2 to competitors, replies to question 36.1. Questionnaire to KOLs, replies to question 9.
53 Annex 7.8 to Form CO.
54 J&J is also developing an oral IL-23R antagonist (JNJ-67864238) which recently started Phase II trials (in September 2019).
55 The fact that all pipelines may not reach the market is illustrated by the Parties’ internal documents, […].
56 Questionnaire Q1 to customers, replies to question 14.2.
57 Questionnaire to KOLs, replies to question 5. For instance, one KOL noted that “it is important as drugs hits an identical target and with biological resistance due to immunogenicity could arise, best example the efficacy of adalimumab in infliximab failures”, another one that “patients who initially respond to a medication may lose response due to antidrug antibody formation and hence have undetectable trough drug levels. an alternative drug in the same class is ideal, as you know that the patient will respond (from previous experience) and that antidrug antibodies do not cross react with agents of the same class (excluding biosimilars of course)”, and an additional KOL “[believes] that it is highly important to have more than one drug within a same class, since this push the competition and differentiation at many levels. This includes, but is not restricted to, aspects such as price, push for RWD (to demonstrate clinical effectiveness and safety beyond the subgroup of patients who goes into a randomized controlled phase III trial) and development of phase IV trials to further differentiate the drug and demonstrate strengths and weaknesses of the drug (including possible head-to-head comparisons)”.
58 On this potential market, the Transaction notably gives rise to other pipeline-to-pipeline overlaps involving upadacitinib (AbbVie), ABBV-323 (AbbVie) and ABI-M201 (Allergan), described in Table 1. KOLs indicated that these three pipeline drugs have very different MoAs (i.e. JAK inhibitor, CD-40 antagonist, and microbiome biologics) compared to the Parties’ other marketed and pipeline drugs for UC and CD. Moreover, Allergan’s ABI-M201 is at an early-stage of development (in Phase I), and thus many years from a hypothetical launch on the market. At this point in time, and based on the available information, there is no indication that the efficacy and safety profiles of these drugs would be similar to other marketed and pipeline drugs of the Parties. See non-confidential minutes of call with KOL dated 18 September 2019, and 16 September 2019. The Transaction is therefore unlikely to raise concerns in relation to these products.
59 Questionnaire Q1 to customers, replies to question 14.1.
60 Questionnaire Q2 to competitors, replies to questions 27.1 to 27.8. Non-confidential minutes of call with KOL dated 16 September 2019, paras. 11 to 15. Non-confidential minutes of call with KOL dated 17 September 2019, paras. 11 to 14. Non-confidential minutes of call with KOL dated 18 September 2019, paras. 14 to 16.
61 Questionnaire Q2 to competitors, replies to question 27.5.
62 A post-marketing study showed an increased risk of pulmonary embolism, malignancy, and overall mortality in rheumatoid arthritis patients. As a result of this safety signal, in July 2019, the FDA approved new warnings about an increased risk of blood clots and death with certain dosages of Xelianz. The implications of this safety signal are currently being discussed by the EMA.
63 Questionnaire Q1 to customers, replies to question 15. Questionnaire Q2 to competitors, replies to questions 27.1 to 27.8. Non-confidential minutes of call with KOL dated 16 September 2019, para. 20. Non-confidential minutes of call with KOL dated 17 September 2019, para. 15. Non-confidential minutes of call with KOL dated 18 September 2019, para. 18: “IL-23 inhibitors are very promising and have the potential to be a game-changer for the treatment of IBD, with a high level of patients achieving clinical remission (40-50%)”.
64 See for instance Annex 7.4 to Form CO.
65 See for instance Questionnaire Q2 to competitors, replies to question 13.
66 See Form CO, Annex 7.6, p. 23: “[…]”. In CD, Allergan is comparing the efficacy and safety of brazikumab with AbbVie’ anti-TNF (Humira) (see Form CO, Annex 7.6, p. 21: “[…]”).
67 Questionnaire R1 to KOLs, replies to question 3.1. See also Questionnaire R2 to Customers, replies to question 3 (a customer stated that “head-to-head trials against existing biologics may allow positioning of brazikumab in the treatment armatorium which is very important information and may give the drug an advantage against other il-23 inhibitors in the case it is found to be superior regarding effect or safety. […] Biomarkers predictive of drug response are important to use drug more selectively and effectively […] This may therefore also give brazikumab a market advantage when it is introduced”); and Questionnaire R3 to Competitors, replies to question 6 (a competitor stated that “it will be very important to show superiority on head-to-head basis for reimbursement purposes […]”; another one indicated that “the comparator studies and a potential indication for a biomarker-defined population would provide differentiation and guidance to prescribers and be positive for market access”).
68 The market-to-pipeline overlap between AbbVie’s Humira and Allergan’s pipeline product ABI-M201 is unlikely to raise concerns given the different MoAs of these products and the very early stage of development of ABI-M201 (Phase I).
69 The pipeline-to-pipeline overlaps involving upadacitinib (AbbVie), ABBV-323 (AbbVie) and ABI-M201 (Allergan) are unlikely to raise concerns, as explained in footnote 58.
70 Biosimilars of adalimumab were approved in the EEA in 2017 and entered the market in 2018.
71 The Parties submit that no reliable market share data is available for Croatia.
72 The Parties submit that no reliable market share data is available for Croatia.
73 Based on data provided until April 2019, Form CO, Annex 7.1.
74 Horizontal Merger Guidelines, paragraph 17.
75 Questionnaire Q2 to competitors, replies to question 6.
76 See Questionnaire Q1 to customers, replies to question 13. One customer notes for instance that “over many years AbbVie has build up experience and have done a good job in building networks and contacts to KOL”, another one that “[l]ong experience with the disease entities, established contact to key opinion leaders, established brand within the field […] would give [AbbVie] some advantages compared to companies with no prior track record”, an additional customer confirms that “[o]ver the years, Abbvie has developed a huge network of contacts in IBD centers over the Europe. IBD specialists have become overly familiar with Abbvie brand and products. For these reasons, Abbvie will have a competitive advantage over all other companies”, similarly another customer considers that “[a]s a company with an esteemed track record in the portfolio of products to treat IBD, any new product with the AbbVie brand must has an advantage in competitive marketing. Probably, the network of the company comprises many peers in the IBD-world, which may also be a competitive advantage”.
77 Questionnaire Q2 to competitors, replies to question 32.1.1.
78 Questionnaire Q2 to competitors, replies to question 15.
79 See Form CO, Annex 7.6, p. 21: “[…]”.
80 J&J’s Simponi is an anti-TNF which is scheduled to lose exclusivity in the EEA in 2024.
81 […].
82 For completeness, Allergan also markets Pred Forte, corticosteroid eye drops indicated for the short-term treatment of eye inflammation. Corticosteroid eye drops are usually the first treatment used for uveitis that affects the front of the eye only (i.e. anterior uveitis). Pred Forte is not therefore substitutable with AbbVie’s Humira, which is not indicated for anterior uveitis.
83 In 2018, Allergan sold Ozurdex in the following EEA countries: […].
84 See Section 3.1 above.
85 Ozurdex is given locally to limit systemic effects via intravitreal implants.
86 See non-confidential minutes of call with KOL dated 26 September 2019 [original wording in French reads: “Humira ne constitue pas une alternative à Ozurdex. Ces deux produits sont utilisés de manière séquencée, dans des cas différents et ne sont pas substituables”].
87 See non-confidential minutes of call with KOL dated 26 September 2019.
88 Commission Notice on remedies acceptable under Council Regulation (EC) No 139/2004 and under Commission Regulation (EC) No 802/2004, the “Remedies Notice” (OJ C 267, 22.10.2008, p. 1-27).
89 Remedies Notice, paragraph 9.
90 Remedies Notice, paragraph 12.
91 Remedies Notice, paragraph 10.
92 Form RM, paragraphs 2.3 to 2.5.
93 Questionnaire R3 to Competitors, replies to questions 4 and 5.
94 Questionnaire R1 to KOLs, replies to question 1; Questionnaire R2 to Customers, replies to question 1; and Questionnaire R3 to Competitors, replies to question 1.
95 Questionnaire R1 to KOLs, replies to question 2; Questionnaire R2 to Customers, replies to question 2; and Questionnaire R3 to Competitors, replies to questions 2 and 3.
96 Questionnaire R1 to KOLs, replies to question 6; Questionnaire R2 to Customers, replies to question 6; and Questionnaire R3 to Competitors, replies to question 10.
97 Questionnaire R3 to Competitors, replies to question 9.
98 Questionnaire R1 to KOLs, replies to question 3 (a KOL stated that “the head-to-head comparison may be used as one of the strongest argument (if not the strongest) for positioning brazikumab […] if the use of IL22 as a companion diagnostic holds true it would probably be rapidly picked up and implemented at IBD-specialised centres […]”); Questionnaire R2 to Customers, replies to question 3 (a customer stated that “head-to-head trials against existing biologics may allow positioning of brazikumab in the treatment armatorium which is very important information and may give the drug an advantage against other il-23 inhibitors in the case it is found to be superior regarding effect or safety. […] Biomarkers predictive of drug response are important to use drug more selectively and effectively […] This may therefore also give brazikumab a market advantage when it is introduced”); and Questionnaire R3 to Competitors, replies to question 6 (a competitor stated that “it will be very important to show superiority on head-tohead basis for reimbursement purposes […]”; another one indicated that “the comparator studies and a potential indication for a biomarker-defined population would provide differentiation and guidance to prescribers and be positive for market access”).
99 Questionnaire R1 to KOLs, replies to questions 9 and 10; Questionnaire R2 to Customers, replies to questions 9 and 10; and Questionnaire R3 to Competitors, replies to questions 14 and 15.
100 Questionnaire R1 to KOLs, replies to question 8; Questionnaire R2 to Customers, replies to questions 7 and 8; and Questionnaire R3 to Competitors, replies to question 12.
CASE NO. COMP/M.9461 – ABBVIE/ALLERGAN
Commitments to the European Commission
Pursuant to Article 6(2) of Council Regulation (EC) No. 139/2004 (the “Merger Regulation”), AbbVie Inc. (“AbbVie”) and Allergan plc (“Allergan”) hereby enter into the following Commitments (the “Commitments”) vis-à-vis the European Commission (the “Commission”) with a view to rendering the acquisition by AbbVie of sole control over Allergan (the “Concentration”) compatible with the internal market and the functioning of the EEA Agreement.
This text shall be interpreted in light of the Commission’s decision pursuant to Article 6(1)(b) of the Merger Regulation to declare the Concentration compatible with the internal market and the functioning of the EEAAgreement (the “Decision”), in the general framework of European Union law, in particular in light of the Merger Regulation, and by reference to the Commission Notice on remedies acceptable under Council Regulation (EC) No 139/2004 and under Commission Regulation (EC) No 802/2004 (the “Remedies Notice”).
SECTION A: DEFINITIONS
1. For the purpose of the Commitments, the following terms shall have the following meaning
AbbVie: AbbVie Inc., a Delaware incorporated company with its registered office at 1 North Waukegan Road, North Chicago, Illinois, 60064-6400.
Affiliated Undertakings: undertakings controlled by the Parties and/or by the ultimate parents of the Parties, whereby the notion of control shall be interpreted pursuant to Article 3 of the Merger Regulation and in light of the Commission Consolidated Jurisdictional Notice under Council Regulation (EC) No 139/2004 on the control of concentrations between undertakings (the “Consolidated Jurisdictional Notice”).
Allergan: Allergan plc, a company incorporated under the laws of Ireland, with its registered office at Clonshaugh Business and Technology Park, Coolock, Dublin, D17 E400, Ireland.
Assets: the assets that contribute to the current operation or are necessary to ensure the viability and competitiveness of the Divestment Business (including those indicated in Section C, paragraph 10 (i), (ii) and (iii)) and described in more detail in the Schedule.
AstraZeneca: AstraZeneca plc, a company incorporated under the laws of England and Wales, with its registered office at 1 Francis Crick Avenue, Cambridge Biomedical Campus, Cambridge, CB2 0AA.
AstraZeneca Agreement: the Licence Agreement between AstraZeneca and Allergan, dated […], and modifications thereto, whereby AstraZeneca grants to Allergan exclusive worldwide patent and know-how rights to develop, manufacture, commercialise and otherwise exploit Brazikumab. These rights are granted by […].
AstraZeneca/Amgen Collaboration: agreement for the joint development, manufacture and commercialisation of various products, including Brazikumab.
Best Efforts: Best effort obligations shall be interpreted in light of the Commission's decision pursuant to Article 6(1)(b) of the Merger Regulation to declare the Concentration compatible with the internal market and the functioning of the EEA Agreement, the Merger Regulation and the general principles of EU law. Any interpretation that may be given to this term under the law of other jurisdictions is not relevant solely for the purpose of interpreting and/or implementing the Commitments.
Brazikumab: Allergan’s in-licensed pipeline IL-23 p19 biologic, previously known as MEDI2070 and AMG139, which is currently in Phase II clinical trials for the treatment of ulcerative colitis ("UC") and Phase IIb / III clinical trials for the treatment of Crohn’s disease ("CD").
Brazikumab drug substance: the active ingredient of Brazikumab.
Closing: the transfer of the legal title to the Divestment Business to thePurchaser.
Closing Period: the period of […] from the approval of the Purchaser and the terms of sale by the Commission.
Confidential Information: any business secrets, know-how, commercial information, or any other information of a proprietary nature that is not in the public domain.
Conflict of Interest: any conflict of interest that impairs the Trustee's objectivity and independence in discharging its duties under the Commitments.
Divestment Business: the development programme and all relevant assets related to Brazikumab as defined in Section B and in the Schedule which the Parties commit to divest.
Divestiture Trustee: one or more natural or legal person(s) who is/are approved by the Commission and appointed by the Parties and who has/have received from the Parties the exclusive Trustee Mandate to sell the Divestment Business to a Purchaser at no minimum price.
Effective Date: the date of adoption of theDecision.
First Divestiture Period: the period of […] from the Effective Date.
Hold Separate Manager: the person appointed by the Parties for the Divestment Business to manage the day-to-day business under the supervision of the MonitoringTrustee.
Key Personnel: all personnel necessary to maintain the viability and competitiveness of the Divestment Business, as listed in Annex 4 to the Commitments, including the Hold Separate Manager.
Monitoring Trustee: one or more natural or legal person(s), who is/are approved by the Commission and appointed by the Parties, and who has/have the duty to monitor the Parties’ compliance with the conditions and obligations attached to theDecision.
Parties: Allergan and AbbVie.
Personnel: all staff currently involved in the development of Brazikumab, as listed in Annex 5 to the Commitments.
Purchaser: entity approved by the Commission as acquirer of the Divestment Business in accordance with the criteria set out in Section D.
Purchaser Agreement: the asset purchase agreement for the sale of the Divestment Business, or other relevant and legally binding agreement(s) required to effect the transfer of the Divestment Business to the Purchaser.
Purchaser Criteria: the criteria laid down in paragraph 18 of these Commitments that the Purchaser must fulfil in order to be approved by theCommission.
Schedule: the schedule to these Commitments describing in more detail the Divestment Business.
Trustee(s): the Monitoring Trustee and/or the Divestiture Trustee as the case may be.
Trustee Divestiture Period: the period of […] from the end of the First Divestiture Period.
SECTION B: THE COMMITMENT TO DIVEST AND THE DIVESTMENT BUSINESS
Commitment to divest
2. In order to maintain effective competition, the Parties commit to divest, or procure the divestiture of, the Divestment Business by the end of the Trustee Divestiture Period as a going concern to a purchaser and on terms of sale approved by the Commission in accordance with the procedure described in paragraphs 19 and 20 of these Commitments. To carry out the divestiture, the Parties commit to find a purchaser and to enter into a final binding Purchaser Agreement within the First Divestiture Period. If the Parties have not entered into such an agreement at the end of the First Divestiture Period, the Parties shall grant the Divestiture Trustee an exclusive mandate to sell the Divestment Business in accordance with the procedure described in paragraph 32 in the Trustee Divestiture Period.
3. The Concentration shall not be implemented before the Parties or the Divestiture Trustee has entered into a final binding Purchaser Agreement (as well as ancillary agreements) and the Commission has approved the Purchaser and the terms of sale in accordance with paragraphs 19 and 20.
4. The Parties shall be deemed to have complied with this commitment if:
(i) by the end of the Trustee Divestiture Period, AbbVie, Allergan, or the Divesture Trustee has entered into a final binding Purchaser Agreement (as well as ancillary agreements), and the Commission approves the proposed purchaser and the terms of transfer or sale as being consistent with the Commitments in accordance with the procedure described in paragraphs 19 and 20; and
(ii) the Closing of the sale of the Divestment Business to the Purchaser takes place within the Closing Period.
5. In order to maintain the structural effect of the Commitments, AbbVie shall, for a period of 10 years after Closing, not acquire, whether directly or indirectly, the possibility of exercising influence (as defined in paragraph 43 of the Remedies Notice, footnote 3) over the whole or part of the Divestment Business, unless, following the submission of a reasoned request from AbbVie showing good cause and accompanied by a report from the Monitoring Trustee (as provided in paragraph 46 of these Commitments), the Commission finds that the structure of the market has changed to such an extent that the absence of influence over the Divestment Business is no longer necessary to render the proposed Concentration compatible with the internal market.
Structure and definition of the Divestment Business
6. The Divestment Business consists of all relevant rights, title and interests in Brazikumab worldwide, including in particular the assignment or transfer to the Purchaser or, (if relevant) termination, of Allergan’s exclusive licence to develop and commercialise Brazikumab in terms of the AstraZeneca Agreement, as well as certain ancillary assets and the rights to, or licence for, certain technologies needed to successfully develop Brazikumab. The legal and functional structure of the Divestment Business as operated to date is described in the Schedule. The Divestment Business, described in more detail in the Schedule, includes all assets and staff that contribute to the current operation of the Divestment Business or that are necessary to ensure the viability and competitiveness of the Divestment Business, in particular:
(i) all tangible and intangible assets (including intellectual property rights);
(ii) all licences, permits and authorisations issued by any governmental organisation for the benefit of the Divestment Business;
(iii) all contracts, and commitments of the Divestment Business and all credit and other records of the Divestment Business;
(iv) the Key Personnel. Allergan will use its Best Efforts to obtain the consent of the Key Personnel to transfer with the Divestment Business. For a period of […] after Closing, the Purchaser shall also have the opportunity to interview the Personnel and to enter into employment contracts with any Personnel.
7. The Divestment Business shall not include any physical production assets owned or operated by the Parties. It will include a small volume of inventory (including samples, work-in-process and pharmaceutical ingredients inventory).
8. The Parties commit to use Best Efforts to ensure an effective transfer of the clinical trials to the Purchaser, as well as reasonable support relating inter alia to regulatory interactions (as reasonably requested by the Purchaser) and assignment of contracts, regulatory permits and licences. Furthermore, the Parties commit to continue to act as a sponsor for all clinical trials underway for the period between the Effective Date until Closing. At the Purchaser’s request, and subject to the Commission’s approval, with respect to any clinical trial, this period can be extended for a mutually agreed period, […].
9. In addition, the Divestment Business includes the benefit, for a transitional period of up to […] after Closing and on terms and conditions equivalent to those at present afforded to the Divestment Business, of all current arrangements under which Allergan or its Affiliated Undertakings supply products or services to the Divestment Business, as detailed in the Schedule, unless otherwise agreed with the Purchaser. Strict firewall procedures will be adopted so as to ensure that any competitively sensitive information related to, or arising from such supply arrangements (for example, product roadmaps) will not be shared with, or passed on to, anyone outside the Parties’ entity(ies) providing such services.
SECTION C: RELATED COMMITMENTS
Preservation of viability, marketability and competitiveness
10. From the Effective Date until Closing, the Parties shall preserve, or procure the preservation of the economic viability, marketability and competitiveness of the Divestment Business, in accordance with good business practice, and shall minimise as far as possible any risk of loss of competitive potential of the Divestment Business. In particular, the Parties undertake:
(i) not to carry out any action that might have a significant adverse impact on the value, management or competitiveness of the Divestment Business or that might alter the nature and scope of activity, or the industrial or commercial strategy or the investment policy of the Divestment Business;
(ii) to make available, or procure to make available, sufficient resources for the development of the Divestment Business, on the basis and continuation of the existing business plans;
(iii) to use Best Efforts, including appropriate incentive schemes (based on industry practice), to encourage all Key Personnel to remain with the Divestment Business, and not to solicit or move any Key Personnel to the Parties’ remaining businesses. Where, nevertheless, individual members of the Key Personnel exceptionally leave the Divestment Business, the Parties shall provide a reasoned proposal to replace the person or persons concerned to the Commission and the Monitoring Trustee. The Parties must be able to demonstrate to the Commission that the replacement is well suited to carry out the functions exercised by those individual members of the Key Personnel. The replacement shall take place under the supervision of the Monitoring Trustee, who shall report to the Commission.
Hold-separate obligations
11. The Parties commit from the Effective Date until Closing to procure that the Divestment Business is kept separate from the business(es) they will be retaining and, after closing of the Concentration, to keep the Divestment Business separate from the business(es) they are retaining and to ensure that unless explicitly permitted under these Commitments:
(i) Management and staff of the business(es) retained by the Parties have no involvement in the Divestment Business;
(ii) Key Personnel of the Divestment Business have no involvement in any business retained by the Parties and do not report to any individual outside the Divestment Business, except for certain Key Personnel to the extent indicated in Annex 4 to the Commitments;
(iii) Personnel of the Divestment Business and the Key Personnel identified in Annex 4 can be involved in businesses retained by the Parties only to the extent that (a) they are bound by the terms of non-disclosure agreements or similar arrangements preventing the disclosure of any information; (b) they are not involved in any UC/CD businesses retained by the Parties; and (c) their involvement in other businesses retained by the Parties is compatible with the required involvement in the Divestment Business.
12. Until Closing, the Parties shall assist the Monitoring Trustee in ensuring that the Divestment Business is managed as a distinct and saleable entity separate from the businesses which the Parties are retaining. Immediately after the adoption of the Decision, the Parties shall appoint a Hold Separate Manager. The Hold Separate Manager, who shall be part of the Key Personnel shall manage the Divestment Business independently and in the best interest of the business with a view to ensuring its continued economic viability, marketability and competitiveness and its independence from the businesses retained by the Parties. The Hold Separate Manager shall closely cooperate with and report to the Monitoring Trustee, and, if applicable, the Divestiture Trustee. Any replacement of the Hold Separate Manager shall be subject to the procedure laid down in paragraph 10(iii) of these Commitments. The Commission may, after having heard the Parties, require the Parties to replace the Hold Separate Manager.
Ring-fencing
13. The Parties shall implement, or procure to implement, all necessary measures to ensure that the Parties do not, after the Effective Date, obtain any Confidential Information relating to the Divestment Business and that any such Confidential Information obtained by the Parties before the Effective Date will be eliminated and not be used by the Parties. In particular, the participation of the Divestment Business in any central information technology network shall be severed to the extent possible, without compromising the viability of the Divestment Business. The Parties may obtain or keep information relating to the Divestment Business which is reasonably necessary for the divestiture of the Divestment Business or the disclosure of which to the Parties is required by law.
Non-solicitation clause
14. The Parties undertake, subject to customary limitations, not to solicit, and to procure that Affiliated Undertakings do not solicit, the Key Personnel and Personnel transferred with the Divestment Business for a period of […] after Closing.
Due diligence
15. In order to enable potential purchasers to carry out a reasonable due diligence of the Divestment Business, the Parties shall, subject to customary confidentiality assurances and dependent on the stage of the divestiture process:
(i) provide to potential purchasers sufficient information as regards the Divestment Business;
(ii) provide to potential purchasers after the Effective Date, a version of the Commitments (including the Schedule and its Annexes) without undue delay and no later than at the signing of a Non-Disclosure Agreement by the potential purchaser, or at the opening a data-room, whichever is earlier. Any redaction to the Commitments should be agreed in advance with the Commission.
(iii) provide to potential purchasers sufficient information relating to the Personnel and allow them reasonable access to the Personnel.
Reporting
16. The Parties shall submit written reports in English on potential purchasers of the Divestment Business and developments in the negotiations with such potential purchasers to the Commission and the Monitoring Trustee no later than 10 days after the end of every month following the Effective Date (or otherwise at the Commission’s request). The Parties shall submit a list of all potential purchasers having expressed interest in acquiring the Divestment Business to the Commission at each and every stage of the divestiture process, as well as a copy of all the offers made by potential purchasers within five days of their receipt.
17. The Parties shall inform the Commission and the Monitoring Trustee on the preparation of the data room documentation and the due diligence procedure and shall submit a copy of any information memorandum to the Commission and the Monitoring Trustee before sending the memorandum out to potential purchasers.
SECTION D: THE PURCHASER
18. In order to be approved by the Commission, the Purchaser must fulfil the following criteria:
(i) The Purchaser shall be independent of and unconnected to the Parties and their Affiliated Undertakings (this being assessed having regard to the situation following the divestiture);
(ii) The Purchaser shall have the financial resources, proven expertise and incentive to maintain and develop the Divestment Business as a viable and active competitive force in competition with the Parties and other competitors. The Purchaser should in particular have the incentive to pursue the clinical trials for the EEA approval of Brazikumab as they are designed as of the Effective Date;
(iii) The acquisition of the Divestment Business by the Purchaser must neither be likely to create, in light of the information available to the Commission, prima facie competition concerns nor give rise to a risk that the implementation of the Commitments will be delayed. In particular, the Purchaser must reasonably be expected to obtain all necessary approvals from the relevant regulatory authorities for the acquisition of the Divestment Business;
(iv) The Purchaser shall have expertise and experience in the clinical development of medicinal products for EEA approval;
(v) The Purchaser shall have expertise and experience in having relevant interactions with relevant EEA-wide and national bodies that decide on approval of medicinal products in the EEA;
(vi) The Purchaser shall have experience in the pricing and reimbursement, marketing, promotion, sales and distribution of medicinal products in the EEA, or the ability to develop such capabilities for the marketing of Brazikumab.
19. The final binding Purchaser Agreement (as well as ancillary agreements) shall be conditional on the Commission’s approval.
20. When Allergan has reached an agreement with a purchaser, the Parties shall submit a fully documented and reasoned proposal, including a copy of the final agreement(s), within one week to the Commission and the Monitoring Trustee. The Parties must be able to demonstrate to the Commission that the Purchaser fulfils the Purchaser Criteria and that the Divestment Business is being sold in a manner consistent with the Commission’s Decision and the Commitments. For the approval, the Commission shall verify that the Purchaser fulfils the Purchaser Criteria and that the Divestment Business is being sold in a manner consistent with the Commitments including their objective to bring about a lasting structural change in the market. The Commission may approve the sale of the Divestment Business without one or more Assets, or parts of the Key Personnel, or by substituting one or more Assets or parts of the Key Personnel with one or more different assets or different personnel, if this does not affect the viability and competitiveness of the Divestment Business after the sale, taking account of the proposed purchaser.
SECTION E TRUSTEE
I. Appointment procedure
21. The Parties shall appoint a Monitoring Trustee to carry out the functions specified in these Commitments for a Monitoring Trustee. The Parties commit not to close the Concentration before the appointment of a Monitoring Trustee.
22. If Allergan has not entered into a binding Purchaser Agreement […] before the end of the First Divestiture Period or if the Commission has rejected a purchaser proposed by the Parties at that time or thereafter, the Parties shall appoint a Divestiture Trustee. The appointment of the Divestiture Trustee shall take effect upon the commencement of the Trustee Divestiture Period.
23. The Trustee shall:
(i) At the time of appointment, be independent of the Parties and their Affiliated Undertakings;
(ii) Possess the necessary qualifications to carry out its mandate, for example have sufficient relevant experience as an investment banker or consultant or auditor; and
(iii) Neither have nor become exposed to a Conflict of Interest.
24. The Trustee shall be remunerated by the Parties in a way that does not impede the independent and effective fulfilment of its mandate. In particular, where the remuneration package of a Divestiture Trustee includes a success premium linked to the final sale value of the Divestment Business, such success premium may only be earned if the divestiture takes place within the Trustee Divestiture Period.
Proposal by the Parties
25. No later than two weeks after the Effective Date, the Parties shall submit the name or names of one or more natural or legal persons whom the Parties propose to appoint as the Monitoring Trustee to the Commission for approval. No later than one month before the end of the First Divestiture Period or on request by the Commission, the Parties shall submit a list of one or more persons whom the Parties propose to appoint as Divestiture Trustee to the Commission for approval. The proposal shall contain sufficient information for the Commission to verify that the person or persons proposed as Trustee fulfil the requirements set out in paragraph 23 and shall include:
(i) the full terms of the proposed mandate, which shall include all provisions necessary to enable the Trustee to fulfil its duties under theseCommitments;
(ii) the outline of a work plan which describes how the Trustee intends to carry out its assigned tasks;
(iii) an indication whether the proposed Trustee is to act as both Monitoring Trustee and Divestiture Trustee or whether different trustees are proposed for the two functions.
Approval or rejection by the Commission
26. The Commission shall have the discretion to approve or reject the proposed Trustee(s) and to approve the proposed mandate subject to any modifications it deems necessary for the Trustee to fulfil its obligations. If only one name is approved, the Parties shall appoint or cause to be appointed the person or persons concerned as Trustee, in accordance with the mandate approved by the Commission. If more than one name is approved, the Parties shall be free to choose the Trustee to be appointed from among the names approved. The Trustee shall be appointed within one week of the Commission’s approval, in accordance with the mandate approved by the Commission.
New proposal by the Parties
27. If all the proposed Trustees are rejected, the Parties shall submit the names of at least two more natural or legal persons within one week of being informed of the rejection, in accordance with paragraphs 21 and 26 of these Commitments.
Trustee nominated by the Commission
28. If all further proposed Trustees are rejected by the Commission, the Commission shall nominate a Trustee, whom the Parties shall appoint, or cause to be appointed, in accordance with a trustee mandate approved by the Commission.
II. Functions of the Trustee
29. The Trustee shall assume its specified duties and obligations in order to ensure compliance with the Commitments. The Commission may, on its own initiative or at the request of the Trustee or the Parties, give any orders or instructions to the Trustee in order to ensure compliance with the conditions and obligations attached to the Decision.
Duties and obligations of the Monitoring Trustee
30. The Monitoring Trustee shall:
(i) propose in its first report to the Commission a detailed work plan describing how it intends to monitor compliance with the obligations and conditions attached to the Decision;
(ii) oversee, in close co-operation with the Hold Separate Manager, the on-going management of the Divestment Business with a view to ensuring its continued economic viability, marketability and competitiveness and monitor compliance by the Parties with the conditions and obligations attached to the Decision. To that end the Monitoring Trustee shall:
(a) monitor the preservation of the economic viability, marketability and competitiveness of the Divestment Business, and the keeping separate of the Divestment Business from the businesses retained by the Parties, in accordance with paragraphs 10 and 11 of these Commitments;
(b) supervise the management of the Divestment Business as a distinct and saleable entity, in accordance with paragraph 12 of these Commitments;
(c) with respect to Confidential Information:
(I) determine all necessary measures to ensure that the Parties do not after the Effective Date obtain any Confidential Information relating to the Divestment Business;
(II) in particular strive for the severing of the Divestment Business’ participation in a central information technology network to the extent possible, without compromising the viability of the Divestment Business;
(III) make sure that any Confidential Information relating to the Divestment Business obtained by the Parties before the Effective Date is eliminated and will not be used by the Parties; and
(IV) decide whether such information may be disclosed to or kept by the Parties as the disclosure is reasonably necessary to allow the Parties to carry out the divestiture or as the disclosure is required by law;
(d) monitor the splitting of assets and the allocation of Personnel between the Divestment Business and the Parties or AffiliatedUndertakings;
(iii) propose to the Parties such measures as the Monitoring Trustee considers necessary to ensure the Parties’ compliance with the conditions and obligations attached to the Decision, in particular the maintenance of the full economic viability, marketability or competitiveness of the Divestment Business, the holding separate of the Divestment Business and the non-disclosure of competitively sensitive information;
(iv) review and assess potential purchasers as well as the progress of the divestiture process and verify that, dependent on the stage of the divestiture process:
(a) potential purchasers receive sufficient and correct information relating to the Divestment Business and the Personnel in particular by reviewing, if available, the data room documentation, the information memorandum and the due diligence process, and
(b) potential purchasers are granted reasonable access to the Personnel;
(v) act as a contact point for any requests by third parties, in particular potential purchasers, in relation to the Commitments;
(vi) provide to the Commission, sending the Parties a non-confidential copy at the same time, a written report within 15 days after the end of every month that shall cover the operation and management of the Divestment Business as well as the splitting of assets and the allocation of Personnel so that the Commission can assess whether the business is held in a manner consistent with the Commitments and the progress of the divestiture process as well as potential purchasers;
(vii) promptly report in writing to the Commission, sending the Parties a nonconfidential copy at the same time, if it concludes on reasonable grounds that the Parties are failing to comply with these Commitments;
(viii) within one week after receipt of the documented proposal referred to in paragraph 20 of these Commitments, submit to the Commission, sending the Parties a non-confidential copy at the same time, a reasoned opinion as to the suitability and independence of the proposed purchaser and the viability of the Divestment Business after the sale and as to whether the Divestment Business is sold in a manner consistent with the conditions and obligations attached to the Decision, in particular, if relevant, whether the sale of the Divestment Business without one or more Assets or not all of the Personnel affects the viability of the Divestment Business after the sale, taking account of the proposed purchaser; and
(ix) assume the other functions assigned to the Monitoring Trustee under the conditions and obligations attached to the Decision.
31. If the Monitoring and Divestiture Trustee are not the same legal or natural persons, the Monitoring Trustee and the Divestiture Trustee shall cooperate closely with each other during and for the purpose of the preparation of the Trustee Divestiture Period in order to facilitate each other's tasks.
Duties and obligations of the Divestiture Trustee
32. Within the Trustee Divestiture Period, the Divestiture Trustee shall sell at no minimum price the Divestment Business to a purchaser, provided that the Commission has approved both the purchaser and the final binding Purchaser Agreement (and ancillary agreements) as in line with the Decision and the Commitments in accordance with paragraphs 18, 19 and 20 of these Commitments. The Divestiture Trustee shall include in the Purchaser Agreement (as well as in any ancillary agreements) such terms and conditions as it considers appropriate for an expedient sale in the Trustee Divestiture Period. In particular, the Divestiture Trustee may include in the Purchaser Agreement such customary representations and warranties and indemnities as are reasonably required to effect the sale. The Divestiture Trustee shall protect the legitimate financial interests of the Parties, subject to AbbVie’s unconditional obligation to divest at no minimum price in the Trustee Divestiture Period.
33. In the Trustee Divestiture Period (or otherwise at the Commission’s request), the Divestiture Trustee shall provide the Commission with a comprehensive monthly report written in English on the progress of the divestiture process. Such reports shall be submitted within 15 days after the end of every month with a simultaneous copy to the Monitoring Trustee and a non-confidential copy to the Parties.
III. Duties and obligations of the Parties
34. The Parties shall provide and shall cause their advisors to provide the Trustee with all such co-operation, assistance and information as the Trustee may reasonably require to perform its tasks. The Trustee shall have full and complete access to any of the Parties or the Divestment Business’s books, records, documents, management or other personnel, facilities, sites and technical information necessary for fulfilling its duties under the Commitments and the Parties and the Divestment Business shall provide the Trustee upon request with copies of any document. The Parties and the Divestment Business shall make available to the Trustee one or more offices on their premises and shall be available for meetings in order to provide the Trustee with all information necessary for the performance of its tasks.
35. The Parties shall provide the Monitoring Trustee with all managerial and administrative support that it may reasonably request on behalf of the management of the Divestment Business. This shall include all administrative support functions relating to the Divestment Business which are currently carried out at headquarters level. The Parties shall provide and shall cause their advisors to provide the Monitoring Trustee, on request, with the information submitted to potential Purchasers, in particular give the Monitoring Trustee access to the data room documentation and all other information granted to potential purchasers in the due diligence procedure. The Parties shall inform the Monitoring Trustee on possible purchasers, submit lists of potential purchasers at each stage of the selection process, including the offers made by potential purchasers at those stages, and keep the Monitoring Trustee informed of all developments in the divestiture process.
36. The Parties shall grant or procure Affiliated Undertakings to grant comprehensive powers of attorney, duly executed, to the Divestiture Trustee to effect the sale (including ancillary agreements), the Closing and all actions and declarations which the Divestiture Trustee considers necessary or appropriate to achieve the sale and the Closing, including the appointment of advisors to assist with the sale process. Upon request of the Divestiture Trustee, the Parties shall cause the documents required for effecting the sale and the Closing to be duly executed.
37. The Parties shall indemnify the Trustee and its employees and agents (each an “Indemnified Party”) and hold each Indemnified Party harmless against, and hereby agrees that an Indemnified Party shall have no liability to the Parties for, any liabilities arising out of the performance of the Trustee’s duties under the Commitments, except to the extent that such liabilities result from the wilful default, recklessness, gross negligence or bad faith of the Trustee, its employees, agents or advisors.
38. At the expense of the Parties, the Trustee may appoint advisors (in particular for corporate finance or legal advice), subject to the Parties’ approval (this approval not to be unreasonably withheld or delayed) if the Trustee considers the appointment of such advisors necessary or appropriate for the performance of its duties and obligations under the Mandate, provided that any fees and other expenses incurred by the Trustee are reasonable. Should the Parties refuse to approve the advisors proposed by the Trustee the Commission may approve the appointment of such advisors instead, after having heard the Parties. Only the Trustee shall be entitled to issue instructions to the advisors. Paragraph 37 of these Commitments shall apply mutatis mutandis. In the Trustee Divestiture Period, the Divestiture Trustee may use advisors who served the Parties during the Divestiture Period if the Divestiture Trustee considers this in the best interest of an expedient sale.
39. The Parties agree that the Commission may share Confidential Information proprietary to the Parties with the Trustee. The Trustee shall not disclose such information and the principles contained in Article 17 (1) and (2) of the Merger Regulation apply mutatis mutandis.
40. The Parties agree that the contact details of the Monitoring Trustee are published on the website of the Commission's Directorate-General for Competition and they shall inform interested third parties, in particular any potential purchasers, of the identity and the tasks of the Monitoring Trustee.
41. For a period of 10 years from the Effective Date the Commission may request all information from the Parties that is reasonably necessary to monitor the effective implementation of these Commitments.
IV. Replacement, discharge and reappointment of the Trustee
42. If the Trustee ceases to perform its functions under the Commitments or for any other good cause, including the exposure of the Trustee to a Conflict of Interest:
(i) the Commission may, after hearing the Trustee and the Parties, require the Parties to replace the Trustee; or
(ii) the Parties may, with the prior approval of the Commission, replace the Trustee.
43. If the Trustee is removed according to paragraph 42 of these Commitments, the Trustee may be required to continue in its function until a new Trustee is in place to whom the Trustee has effected a full hand over of all relevant information. The new Trustee shall be appointed in accordance with the procedure referred to in paragraphs 21-28 of these Commitments.
44. Unless removed according to paragraph 42 of these Commitments, the Trustee shall cease to act as Trustee only after the Commission has discharged it from its duties after all the Commitments with which the Trustee has been entrusted have been implemented. However, the Commission may at any time require the reappointment of the Monitoring Trustee if it subsequently appears that the relevant remedies might not have been fully and properly implemented.
SECTION F: THE REVIEW CLAUSE
45. The Commission may extend the time periods foreseen in the Commitments in response to a request from the Parties or, in appropriate cases, on its own initiative. Where the Parties request an extension of a time period, they shall submit a reasoned request to the Commission no later than one month before the expiry of that period, showing good cause. This request shall be accompanied by a report from the Monitoring Trustee, who shall, at the same time send a non-confidential copy of the report to the Parties. Only in exceptional circumstances shall the Parties be entitled to request an extension within the last month of any period.
46. The Commission may further, in response to a reasoned request from the Parties showing good cause waive, modify or substitute, in exceptional circumstances, one or more of the undertakings in these Commitments. This request shall be accompanied by a report from the Monitoring Trustee, who shall, at the same time send a non-confidential copy of the report to the Parties. The request shall not have the effect of suspending the application of the undertaking and, in particular, of suspending the expiry of any time period in which the undertaking has to be complied with.
SECTION G: ENTRY INTO FORCE
47. The Commitments shall take effect upon the date of adoption of the Decision.
(Signed)
Duly authorised for and on behalf of: AbbVie Inc.
Duly authorised for and on behalf of: Allergan plc
Schedule
1. The Divestment Business consists of all relevant rights, title and interests in Brazikumab worldwide including in particular the assignment or transfer to the Purchaser or, (if relevant) termination, of Allergan’s exclusive licence to develop and commercialise Brazikumab in terms of the AstraZeneca Agreement, as well as certain ancillary assets and the rights to or licence for certain technologies needed to successfully develop Brazikumab. The current development programme and ongoing clinical trials for Brazikumab are described in Annex 6.
2. In accordance with Section B, paragraph 6 of these Commitments, the Divestment Business includes but is not limited to:
(i) The following main tangible assets (together with the intangible assets listed below the “TransferredAssets”):
(a) All finished goods inventory, together with any other inventory (including work-in-process, samples and active pharmaceutical ingredients inventory) of Brazikumab that is owned by and in the possession or control, or otherwise held by or on behalf of Allergan or Affiliated Undertakings;
(b) All relevant reports, databases and analysis related to Brazikumab, including all technical, preclinical, clinical and marketing files, protocols, clinical data and studies, reports, plans, books and records relating to Brazikumab. The reports, databases and analysis relating to Brazikumab that also relate to other products developed or to be developed by Allergan shall only be transferred to the extent that they relate to Brazikumab, it being understood that the other sections shall be redacted prior to the transfer to the Purchaser. If required by law, any individual patient data contained in such reports, databases and analysis shall be redacted prior to transfer to the Purchaser;
(c) All original documents or, to the extent original documents are not reasonably available, copies thereof, in any format in the possession or control of Allergan or Affiliated Undertakings as at Closing, (i) relating to all relevant regulatory files, including all Regulatory Approvals (defined below); (ii) of all data that resulted from any research or development activities related to Brazikumab and conducted by or on behalf of Allergan or Affiliated Undertakings or a previous sponsor of an investigational new drug application (“IND”) for Brazikumab; and (iii) correspondence with relevant regulatory bodies relating to the research, development, manufacture, testing, storage, import, export, use, labelling, distribution, sale, offer for sale, commercialisation, licensing, advertising, marketing and promotion (“exploitation”) of Brazikumab (including all INDs and foreign counterparts thereof, periodic safety reports and adverse drug experience reports for Brazikumab);
(d) All documents related to marketing plans and forecasts which are specific to Brazikumab. The marketing plans and forecasts that also relate to other products developed or to be developed by Allergan or its Affiliates shall be transferred only to the extent that they relate to Brazikumab, it being understood that the other sections shall be redacted prior to the transfer to the Purchaser.
(ii) The following main intangible assets:
(a) All intellectual property rights licensed to Allergan under and in accordance with the terms of the AstraZeneca Agreement;
(b) […] pending patent applications and […] application listed in Annex 1;
(c) The rights to conduct the clinical trials sponsored by Allergan or its Affiliated Undertakings and related to Brazikumab globally;
(d) The rights to manufacture (or have manufactured) and market Brazikumab globally;
(e) All other intellectual property rights (including know-how and patents) existing as at the Closing Date that relate exclusively or predominantly to the development, manufacture and/or sale of Brazikumab worldwide, subject to a non-exclusive, perpetual, irrevocable, royalty-free licenceback to the Parties of any such intellectual property rights that are shared with any of Allergan’s retained businesses. Intellectual property rights that are used by or relevant for the Divestment Business, but that are not predominantly related to the development, manufacture and/or sale of Brazikumab, will be subject to a non-exclusive, perpetual, irrevocable, royalty-free licence to the benefit of the Purchaser;
(f) The proposed “brazikumab” International Nonproprietary Name (“INN”), including any relevant registered domain names.
(iii) The following main licences, permits and authorisations (“Regulatory Approvals”): Brazikumab IND number […] and any new INDs, clinical trial authorisations (“CTAs”) or any other authorisations, permits or regulatory approvals (including any pending regulatory approvals) necessary for the exploitation of Brazikumab and held by Allergan or Affiliated Undertakings (as listed in Annex 2).
(iv) All contracts with third parties to which Allergan or Affiliated Undertakings are bound that are primarily related to the exploitation of Brazikumab. A list of the third party contracts is provided in Annex 3. These contracts will transfer to the Purchaser subject to any required consents being obtained. The Parties undertake to use Best Efforts to obtain all necessary third-party consents where applicable. To the extent any such third-party consent could not be obtained, or any contract could not be otherwise transferred, the Parties will, as appropriate, either: (i) assist the Purchaser and the relevant third party to put in place arrangements to transfer any work product and work in progress, and support the Purchaser to put in place alternative arrangements; or (ii) enter into back-toback agreements with the Purchaser under the same terms and conditions as the relevant contract for a transitional period of up to […].
(v) The Key Personnel listed in Annex 4. The Key Personnel will transfer to the Purchaser with the Divestment Business subject to their consent, which the Parties shall use Best Efforts to obtain.
(vi) In respect of the Personnel listed in Annex 5, for a period of […] after Closing, the Purchaser shall have the opportunity to interview, and enter into employment contracts with, any Personnel.
(vii) At the Purchaser’s request, for a period of up to […] from the Closing Date, the Parties will provide, on terms and conditions equivalent to those at present afforded to the Divestment Business, all products or services currently supplied by Allergan or its Affiliated Undertakings to the Divestment Business.
(viii) For a period of up to […] from the Closing Date, the Parties will provide, […], any transitional support required by the Purchaser to enable the continuity of the clinical trials, including providing technical support, liaising with regulatory authorities and the transfer of relevant contracts, licences and CTAs.
3. In the event that materials to be transferred contain information that is confidential to the Parties’ retained businesses and not relevant for the Divestment Business, the information shall be redacted as appropriate.
4. The Divestment Business shall not include the following assets, to the extent that they do not contribute to the current operation or are not necessary to ensure the viability and competitiveness of the Divestment Business:
(i) any trademarks that Allergan or its Affiliated Undertakings own or have the right to use and licence, other than as described in paragraph 2 of this Schedule;
(ii) any contracts with third parties to which Allergan or Affiliated Undertakings are bound, other than those listed in Annex 3;
(iii) any intellectual property (including patents, trademarks and trade names, domain name registrations, copyrights and trade secrets and know how) owned by Allergan or its Affiliated Undertakings, other than as described in paragraph 2 of this Schedule;
(iv) the corporate books and records of Allergan or its Affiliated Undertakings other than as described in paragraph 2 of this Schedule;
(v) any right to manufacture, market or sell any product other than Brazikumab or any licence to use any asset of Allergan in connection with any product other than Brazikumab;
(vi) any current and prior insurance policies and all related rights, including all insurance recoveries and related rights.
5. If there is any asset or personnel which is not covered by paragraph 2 of this Schedule but which is both used (exclusively or not) for the Divestment Business and necessary for the continued viability and competitiveness of the Divestment Business, that asset or adequate substitute will be offered to potential purchasers by transfer or (nonexclusive, perpetual, irrevocable, and royalty-free) licence, as appropriate, as overseen by the Monitoring Trustee.
Annex 1
Patent applications
[...]
Annex 2
Regulatory Approvals
Table 1
Clinical trial authorisations
[...]
Table 2
[…] investigational new drug applications
[…]
Annex 3
Transferring contracts
[...]
Annex 4
Key Personnel
[...]
Annex 5
Personnel
[...]
Annex 6
Brazikumab current development programme and ongoing clinical trials
1. The Divestment Business currently forms part of Allergan’s GI business. Brazikumab was initially developed by Amgen Inc. (“Amgen”). In 2012, Amgen and AstraZeneca entered into the AstraZeneca/Amgen Collaboration to jointly develop, manufacture and commercialise various products, including Brazikumab. […]. Following the conclusion of the AstraZeneca Agreement in […], Allergan now holds the exclusive licence to develop and commercialise brazikumab worldwide at its own cost. […].
2. Brazikumab is currently in Phase II clinical trials for the treatment of moderate to severe ulcerative colitis (“UC”) and in Phase IIb/III trials for the treatment of moderate to severe Crohn’s disease (“CD”). There is also an ongoing 52 week open label study for the treatment of moderate to severe CD.
3. The clinical trials are carried out on at third party locations in the United States, and will be carried out at third party locations on a global basis including in Canada and the EEA. Allergan’s role is to act as a sponsor. […]. Allergan has appointed […] to conduct recruitment and site management services for […]. Allergan also contracts with […] to perform activities such as site selection, site visits and site management for the Brazikumab studies in India and China.
4. With the exception of some packaging and labelling services […], Allergan outsources all other services connected with the running of the clinical trials.
5. […] third party manufacturers – […] –- currently manufacture the Brazikumab drug substance. […].
6. Allergan currently tests brazikumab with an IL-22 companion diagnostic in order to assess whether brazikumab is more effective than existing approved products in treating UC and CD in patients expressing higher levels of the IL-22 biomarker. Any agreements with third parties entered into in relation to companion diagnostics in advance of Closing will be assigned to the Purchaser.
7. […]. Any agreements with third parties entered into in relation to […] in advance of Closing will be assigned to the Purchaser.
8. The table below lists the current ongoing and planned clinical trials for brazikumab.