
ADLC, 9 septembre 2020, n° 20-D-11
AUTORITÉ DE LA CONCURRENCE
Décision
relative à des pratiques mises en œuvre dans le secteur du traitement de la dégénérescence maculaire liée à l’âge (DMLA)
COMPOSITION DE LA JURIDICTION
Délibéré sur le rapport oral de M. Alexis Brunelle, rapporteur, et l’intervention de M. Umberto Berkani, rapporteur général adjoint, par Mme Isabelle de Silva, présidente, Mme Fabienne Siredey-Garnier, vice-présidente, Mme Laurence Borrel-Prat, M. Savinien Grignon-Dumoulin, M. Jean-Yves Mano et M. Christophe Strassel, membres.
L’Autorité de la concurrence (section IB),
Vu la décision n° 15-SO-12 du 17 septembre 2015, enregistrée sous le numéro 15/0083F, par laquelle l’Autorité de la concurrence s’est saisie d’office de pratiques mises en œuvre dans le secteur du traitement de la dégénérescence maculaire liée à l’âge (DMLA) ;
Vu l’article 102 du Traité sur le fonctionnement de l’Union européenne ;
Vu le livre IV du code de commerce, et notamment son article L. 420-2 ;
Vu les décisions de secret d'affaires n° 18-DSA-191 du 15 juin 2018, n° 18-DSA-254 du 06 août 2018, n° 18-DEC-393 du 14 novembre 2018, n° 18-DSA-196 du 18 juin 2018, n° 19-DECR-080 du 26 février 2019, n° 16-DSA-27 du 29 février 2016, n° 16-DSA-28 du 29 février 2016, n° 19-DSA-075 du 27 février 2019, n° 19-DEC-006 du 09 janvier 2019, n° 19-DECR-536 du 26 septembre 2019, n° 19-DEC-021 du 16 octobre 2018, n° 18-DSA-209 du 09 juillet 2018, n° 18-DSA-142 du 23 mai 2018, n° 18-DSA-210 du 10 juillet 2018, n° 18-DSA-222 du 16 juillet 2018, n° 18-DSA-227 du 16 juillet 2018, n° 18-DSA-238 du 26 juillet 2018, n° 19-DSA-143 du 17 mai 2019, n° 19-DSA-185 du 24 mai 2019, n° 19-DSA-249 du 12 juillet 2019, n° 19-DECR-251 du 12 juillet 2019, n° 19-DECR-253 du 12 juillet 2019, n° 19-DSA-711 du 11 décembre 2019, n° 19-DSA-712 du 11 décembre 2019 ;
Vu les autres pièces du dossier ;
Vu les observations présentées par les sociétés Novartis Pharma SAS, Novartis Groupe France SA, Novartis AG, Roche SAS, Genentech Inc., Roche Holding AG et le commissaire du Gouvernement ;
Le rapporteur, le rapporteur général adjoint, les représentants de la direction générale de la santé (pour le ministre de la santé), les représentants des sociétés Novartis Pharma SAS, Novartis Groupe France SA, Novartis AG, Roche SAS, Genentech Inc. et Roche Holding AG, et le commissaire du Gouvernement entendus lors de la séance de l’Autorité de la concurrence du 12 mars 2020 ;
Résumé(1)
Aux termes de la décision ci-après, plusieurs sociétés du groupe Novartis (Novartis Pharma SAS, Novartis Groupe France SA et Novartis AG, ci-après « Novartis ») et du groupe Roche (Roche SAS, Genentech Inc. et Roche Holding AG, ci-après « Roche », ou « Genentech » ou « Roche/Genentech »), ont été sanctionnées à hauteur de 444 millions d’euros, pour avoir abusé de leur position dominante collective sur le marché français du traitement de la DMLA exsudative, en mettant en œuvre plusieurs pratiques contraires aux articles L. 420-2 du code de commerce et 102 du traité sur le fonctionnement de l’Union européenne. L’affaire a été instruite par l’Autorité de la concurrence (ci-après, « l’Autorité ») à la suite d’un signalement transmis par la brigade interrégionale des enquêtes de concurrence (ci-après « BIEC ») de Lyon.
L’Autorité a constaté que Novartis, Roche et Genentech formaient une entité collective, pour les besoins de la commercialisation de Lucentis et Avastin, détenant une position dominante sur le marché du traitement de la DLMA exsudative, compte tenu d’une part, de l’existence de liens structurels importants et stratégiques entre les laboratoires (en particulier les contrats de licence liant Genentech et Novartis, pour la commercialisation de Lucentis, et Genentech et Roche, pour la commercialisation d’Avastin) et, d’autre part, de l’existence de liens capitalistiques croisés entre Genentech, Roche et Novartis. La structure contractuelle et capitalistique existant entre Genentech, Roche et Novartis a permis à ces derniers d’adopter une ligne d’action commune sur le marché concerné, visant à limiter les prescriptions d’Avastin « hors AMM » en ophtalmologie. En particulier, l’Autorité a constaté qu’il existait une forte incitation financière pour les trois laboratoires à adopter une ligne de conduite commune, dont la mise en œuvre a été facilitée par les liens structurels. En effet, compte tenu des différences de prix entre les deux spécialités et de la pratique consistant à fabriquer plusieurs seringues avec un seul flacon d’Avastin, toute utilisation d’Avastin à la place de Lucentis pour une injection dans l’œil était susceptible d’entraîner un manque à gagner significatif pour chacun des trois laboratoires concernés : (i) pour Novartis, d’abord, qui reçoit, en tant que licencié, le produit des ventes de Lucentis sur le marché concerné, (ii) pour Genentech, ensuite, qui perçoit, en tant que donneur de licence, les redevances des ventes de Lucentis sur le marché concerné, et (iii) pour Roche, enfin, qui, en tant qu’actionnaire principal puis unique depuis mars 2009 de Genentech, profite des bénéfices du laboratoire américain.
Novartis a été sanctionné au titre du grief n° 1 pour avoir diffusé, en s’appuyant sur la position dominante collective détenue avec Roche et Genentech sur le marché du traitement de la DMLA, un discours dénigrant, en exagérant, de manière injustifiée, les risques liés à l’utilisation d’Avastin « hors AMM » pour le traitement de la DMLA, et plus généralement en ophtalmologie, en comparaison avec la sécurité et la tolérance de Lucentis pour un même usage. Cette pratique a été de nature à avoir et a eu pour effet de limiter les prescriptions d’Avastin « hors AMM » pour le traitement de la DMLA et, plus généralement en ophtalmologie. Elle est également susceptible d’avoir eu pour effet le maintien de Lucentis à un prix supra-concurrentiel ou encore la fixation du prix d’Eylea, spécialité concurrente arrivée sur le marché français à la fin de l’année 2013, à un niveau artificiellement élevé. Elle a débuté le 10 mars 2008 pour prendre fin début novembre 2013, avec l’arrivée sur le marché de la spécialité concurrente Eylea, commercialisée par le laboratoire Bayer en Europe, qui a mis fin à la position dominante de l’entité collective.
Novartis, Roche et Genentech ont été sanctionnés au titre du grief n° 2 pour avoir diffusé, en s’appuyant sur la position dominante collective détenue par les trois laboratoires sur le marché du traitement de la DMLA, et plus généralement en ophtalmologie, un discours alarmiste, voire trompeur, sur les risques liés à l’utilisation d’Avastin pour le traitement de la DMLA, afin de bloquer ou ralentir, de façon indue, les initiatives des pouvoirs publics qui envisageaient de favoriser et sécuriser son usage pour le traitement de la DMLA. Plus spécifiquement, Genentech a pris part aux pratiques, dans la mesure où il a permis de coordonner, notamment concernant les données scientifiques, le discours de Novartis et de Roche concernant les deux spécialités. Dans ce contexte, Roche et Novartis ont pu développer un discours très similaire, fondé sur les mêmes éléments techniques, fournis en grande partie par Genentech, auprès des autorités de santé. L’infraction a été de nature à décourager les autorités publiques de favoriser un plus large recours « hors AMM » d’Avastin dans le traitement de la DMLA et, par voie de conséquence, à limiter les prescriptions d’Avastin « hors AMM » pour le traitement de la DMLA et, plus généralement en ophtalmologie. Elle est également susceptible d’avoir eu pour effet le maintien de Lucentis à un prix supra-concurrentiel ou encore la fixation du prix d’Eylea à un niveau artificiellement élevé. Elle a débuté le 7 avril 2008 concernant Roche, le 28 avril 2011 concernant Genentech et le 9 mai 2011 concernant Novartis et a pris fin début novembre 2013, avec l’arrivée sur le marché de la spécialité concurrente Eylea, commercialisée en Europe par le laboratoire Bayer, qui a mis fin à la position dominante de l’entité collective.
Pour déterminer le montant des sanctions prononcées, l’Autorité a notamment pris en compte la gravité des pratiques en cause et le dommage certain qu’elles ont causé à l’économie. En particulier, les comportements des laboratoires sanctionnés au titre des deux griefs sont intervenus dans le secteur de la santé où la concurrence est limitée, et plus spécifiquement dans un contexte de débat public sur le prix extrêmement élevé de Lucentis et sur son impact sur les finances sociales, pour lesquelles le remboursement à 100 % de Lucentis constituait un poste de dépense significatif, alors qu’il existait un médicament, Avastin, nettement moins cher, susceptible d’être utilisé en ophtalmologie.
I. Constatations
1. Seront successivement présentés la procédure (A), le secteur d’activité (B), les acteurs concernés (C), l’affaire italienne (D), les pratiques constatées (E) et les griefs notifiés (F).
A. LA PROCEDURE
2. À la suite d’un signalement transmis par la brigade interrégionale des enquêtes de concurrence (ci-après, « BIEC ») de Lyon, l’Autorité de la concurrence (ci-après, « l’Autorité ») a réalisé, le 8 avril 2014, des opérations de visite et saisie dans les locaux des sociétés Novartis Groupe France et Roche, autorisées par ordonnance du juge des libertés et de la détention du Tribunal de grande instance de Nanterre en date du 1er avril 2014. Le déroulement des opérations de visite et saisies a fait l’objet d’un recours(2).
3. Par décision n° 15-SO-12 du 17 septembre 2015, l’Autorité s’est saisie d’office de pratiques mises en œuvre dans le secteur du traitement de la dégénérescence maculaire liée à l’âge (ci-après, « DMLA »).
4. Le 23 janvier 2019, le rapporteur général de l’Autorité a adressé une notification de griefs portant sur des pratiques prohibées au titre de l’article 102 du Traité sur le fonctionnement de l’Union européenne (ci-après, « TFUE ») et de l’article L. 420-2 du code de commerce aux sociétés Novartis Pharma SAS, Novartis Groupe France SA, Novartis AG (ci-après, ensemble, « Novartis »), Genentech Inc. (ci-après, « Genentech ») et Roche SAS et Roche Holding AG (ci-après, ensemble, « Roche »).
B. LE SECTEUR D’ACTIVITE
5. Seront successivement examinés le cadre légal et réglementaire de la commercialisation des médicaments concernés en France (1), les produits concernés par les pratiques (2) et le cadre scientifique concernant l’utilisation d’Avastin dans le traitement des pathologies oculaires (3).
1. LE CADRE LEGAL ET REGLEMENTAIRE DE LA COMMERCIALISATION DES MEDICAMENTS CONCERNES EN FRANCE
6. Il convient de revenir sur l’encadrement de la mise sur le marché d’un médicament (a) et les modalités de fixation de prix des médicaments (b) en France.
a) L’encadrement de la mise sur le marché d’un médicament
La délivrance d’une autorisation de mise sur le marché (ci-après, « AMM ») pour une ou plusieurs indications
Le principe d’obligation d’obtention d’une AMM pour une indication thérapeutique donnée dans l’Union européenne
7. Tout médicament fabriqué industriellement et mis en vente sur un marché national ou dans l'Union européenne doit faire l'objet d'une AMM (article 6, paragraphe 1, de la directive n° 2001/83/CE du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain).
8. Les procédures d’octroi d’AMM permettent d'évaluer les médicaments concernés, notamment en termes de sécurité ou d'efficacité. Les autorités de santé compétentes pour délivrer l'AMM s'appuient sur les résultats d'études effectuées par les laboratoires pharmaceutiques demandeurs de l’autorisation (essais cliniques et précliniques s’agissant des médicaments princeps). Elles ne procèdent, en principe, pas elles-mêmes aux expériences scientifiques requises pour l'analyse des effets d'un médicament.
9. Sur la base de ces études scientifiques, les autorités de santé compétentes réalisent une évaluation du profil bénéfice/risque de la spécialité concernée, concernant la ou les indication(s) thérapeutique(s) envisagée(s). Ce n’est que dans l’hypothèse où la balance bénéfices/risques est favorable que l’autorité de santé délivrera une AMM pour l’indication visée. Dans l’hypothèse d’une évolution de la ou des indication(s) thérapeutique(s) de la spécialité concernée, une nouvelle autorisation devra être délivrée.
La possibilité de dérogation à l’utilisation ciblée d’une spécialité autorisée à être commercialisée
10. L’octroi d’une AMM correspond à l’autorisation de commercialisation d’une spécialité pour une ou plusieurs indication(s) thérapeutique(s). La délivrance d’une AMM pour des indications spécifiques ne fait toutefois pas obstacle à la prescription par les médecins de cette spécialité pour d’autres indications : le cadre communautaire applicable prévoit ainsi la possibilité de prescrire un médicament « hors AMM », c’est-à-dire pour d’autres indications que celles pour lesquelles le médicament a reçu son AMM.
11. L’article 5, paragraphe 1, de la directive n° 2001/83/CE prévoit ainsi : « Un Etat membre peut, conformément à la législation en vigueur et en vue de répondre à des besoins spéciaux, exclure des dispositions de la présente directive les médicaments fournis pour répondre à une commande loyale et non sollicitée, élaborés conformément aux spécifications d’un professionnel de santé agréé et destinés à ses malades particuliers sous sa responsabilité personnelle directe ».
12. Ces dispositions doivent être interprétées de façon stricte (arrêt de la Cour de justice de l’Union européenne du 29 mars 2012, Commission c/ République de Pologne, C-185/10, point 31). Selon la Cour de justice, « la dérogation prévue par cette disposition ne peut concerner que des situations dans lesquelles le médecin estime que l’état de santé de ses patients particuliers requiert l’administration d’un médicament dont il n’existe pas d’équivalent autorisé sur le marché national ou qui se trouve indisponible sur le marché » (point 36). Par ailleurs, « des considérations financières ne sauraient, à elles seules, conduire à reconnaître l’existence de tels besoins spéciaux » (point 38).
13. Concernant spécifiquement l’élaboration de seringues prêtes à l’emploi d’Avastin, la Cour de justice a jugé qu’ « un médecin peut, face à une pathologie donnée et en se fondant uniquement sur des considérations thérapeutiques propres à ses patients, y compris au regard des modalités d’administration du médicament, estimer qu’un traitement hors AMM, selon la forme galénique(3) et la posologie qu’il estime appropriée et au moyen de l’Avastin disposant d’une AMM, communautaire, est préférable à un traitement au moyen du Lucentis » (arrêt du 11 avril 2013, Novartis Pharma GmbH c/ Apozyt GmbH, C-535/11, point 48). De même, elle a considéré que « l’article 6 de la directive 2001/83 doit être interprété en ce sens qu’il ne s’oppose pas à des mesures nationales telles que celles en cause au principal qui définissent les conditions dans lesquelles l’Avastin peut être reconditionné aux fins de son utilisation pour le traitement d’indications ophtalmologiques non couvertes par son AMM » (arrêt du 21 novembre 2018, Novartis c/ AIFA, C-29/17, point 79).
14. Ainsi, la règlementation de l’Union en matière de produits pharmaceutiques n’interdit ni la prescription d’un médicament « hors AMM » ni son reconditionnement en vue d’une telle utilisation, mais les subordonne au respect de certaines conditions (arrêt de la Cour de justice du 23 janvier 2018, F. Hoffmann-La Roche e.a., C-179/16, point 59).
La liberté de prescription du médecin
15. Cette possibilité de prescription « hors AMM » s’inscrit dans le cadre d’un principe bien établi et figurant au nombre des principes déontologiques fondamentaux applicables à la profession médicale, le principe de liberté de prescription.
16. Le principe de la liberté de prescription du médecin est inscrit à l’article L. 162-2 du code de la sécurité sociale, qui dispose « Dans l'intérêt des assurés sociaux et de la santé publique, le respect de la liberté d'exercice et de l'indépendance professionnelle et morale des médecins est assuré conformément aux principes déontologiques fondamentaux que sont le libre choix du médecin par le malade, la liberté de prescription du médecin, le secret professionnel, le paiement direct des honoraires par le malade, la liberté d'installation du médecin (…) ». L’article L. 162-4 du même code fait obligation pour le prescripteur de signaler sur l’ordonnance, support de la prescription, le caractère non remboursable du médicament lorsqu’il l’a prescrit en dehors des indications thérapeutiques ouvrant droit au remboursement ou à la prise en charge par l’assurance maladie.
17. Le code de déontologie médicale rappelle également la portée du principe, en vertu duquel les médecins ont l’obligation légale de délivrer le traitement le plus adapté à chaque patient, en tenant compte des données acquises de la science (article 8 du code de déontologie médicale, codifié à l’article R. 4127-8 du code de la santé publique : « Dans les limites fixées par la loi et compte tenu des données acquises de la science, le médecin est libre de ses prescriptions qui seront celles qu'il estime les plus appropriées en la circonstance »).
18. Ainsi, les médecins ont la possibilité, lorsqu’ils l’estiment nécessaire, de prescrire à leurs patients une spécialité bénéficiant d’une AMM, en dehors de son indication (voir par exemple, explicitant ce point, les arrêts du Conseil d’État du 19 octobre 2001, D…, n° 210590 et de la Cour de cassation du 4 janvier 2005, n° 03-14206 et du 18 septembre 2008, n° 07-15427).
19. Il est à noter que la pratique de prescription « hors AMM » est relativement répandue. Ainsi, ce type de prescription représentait à l’époque des pratiques en cause (en 2011), environ 15 à 20 % de la totalité des prescriptions, tous domaines confondus(4).
L’évolution de la réglementation française concernant les prescriptions « hors AMM »
20. Jusqu’à la fin de l’année 2011, les prescriptions « hors AMM » relevaient de différents régimes. Certains usages « hors AMM » résultaient de recommandations de prescription émises par les autorités sanitaires, d’autres étaient prévus par les textes. Tel était le cas s’agissant des autorisations temporaires d’utilisation (ci-après « ATU »)(5) destinées à soigner des maladies rares avant l’octroi d’une AMM, des protocoles thérapeutiques temporaires (ci-après, « PTT »)(6), ou encore de certains médicaments prescrits pour le traitement ambulatoire des maladies rares et des affections de longue durée (rapport de la Commission des lois sur le projet de loi relatif au renforcement de la sécurité sanitaire du médicament et des produits de santé, septembre 2011, page 137).
21. En réaction à l’affaire du Médiator, révélée en 2009 à la suite de l’utilisation par des médecins d’un médicament antidiabétique prescrit, en dehors de l’indication prévue par son AMM, comme « coupe-faim » à des patients souhaitant perdre du poids, le législateur a décidé, non pas d’interdire les prescriptions « hors AMM » – ce qui aurait non seulement été contraire à la liberté de prescription mais aurait également privé les patients de certains traitements – mais de mieux les encadrer (rapport susvisé, page 138) afin de prévenir les risques pour la santé des patients.
22. Ainsi, la loi n° 2011-202 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé (dite « loi Bertrand », du nom du ministre de la santé de l’époque, Xavier Bertrand) a inséré dans le code de la santé publique un article L. 5121-12-1, créant une nouvelle catégorie juridique pour l’utilisation de médicaments « hors AMM », les recommandations temporaires d’utilisation (ci-après, « RTU »), rédigé comme suit : « I. Une spécialité pharmaceutique peut faire l'objet d'une prescription non conforme à son autorisation de mise sur le marché en l'absence d'alternative médicamenteuse appropriée disposant d'une autorisation de mise sur le marché ou d'une autorisation temporaire d'utilisation, sous réserve : 1° Que l'indication ou les conditions d'utilisation considérées aient fait l'objet d'une recommandation temporaire d'utilisation établie par l'Agence nationale de sécurité du médicament et des produits de santé, cette recommandation ne pouvant excéder trois ans ; 2° Ou que le prescripteur juge indispensable, au regard des données acquises de la science, le recours à cette spécialité pour améliorer ou stabiliser l'état clinique du patient » (soulignements ajoutés).
23. Saisi au contentieux de la légalité de la RTU adoptée en juin 2015 pour autoriser l’usage d’Avastin en dehors de son AMM (cf. paragraphe 72 ci-dessous), le Conseil d’État a précisé la portée de ces dispositions. Ainsi, dans son arrêt du 24 février 2017, portant sur le recours dirigé contre la RTU Avastin, il a souligné sur ce point : « Considérant que, eu égard au développement de la pratique de prescriptions de certaines spécialités en dehors des indications ou des conditions d'utilisation de leur autorisation de mise sur le marché, aux bénéfices susceptibles d'en être attendus ainsi qu'aux risques courus, le législateur a entendu, par l'élaboration de recommandations temporaires d'utilisation, et sans que le deuxième alinéa du I de l'article L. 5121-12-1 du code de la santé publique interdise ces prescriptions en l'absence de recommandation, renforcer les garanties associées à cette pratique par la mise à disposition des médecins, par l'Agence nationale de sécurité du médicament et des produits de santé chargée de leur élaboration, d'informations relatives notamment aux bénéfices attendus de la spécialité et aux risques courus dans l'indication ou les conditions d'utilisation en cause et par la mise en place d'un suivi des patients » (soulignement ajouté).
24. Ainsi, après l’entrée en vigueur de la loi Bertrand, un médecin pouvait toujours décider de prescrire un médicament en dehors de son AMM, dès lors qu’il estimait que le recours à cette spécialité était le plus indiqué pour soigner son patient.
25. L’article L. 5121-12-1 du code de la santé publique a par la suite été modifié, afin de tenir compte du fait que certains laboratoires faisaient le choix commercial ou industriel de ne pas développer une spécialité dans une indication donnée. Le législateur a donc décidé d’étendre la possibilité d’adopter, à titre exceptionnel, une RTU pour une spécialité pharmaceutique dans des cas où il existe une alternative thérapeutique dans cette indication, en réponse soit à un impératif de santé publique, soit à une exigence de maîtrise des dépenses de l’assurance maladie (étude d’impact relative au projet de loi de financement de la sécurité sociale pour 2013, octobre 2012, page 231).
26. Ainsi, la loi n° 2012-1404 du 17 décembre 2012 de financement de la sécurité sociale pour 2013 (ci-après, « LFSS 2013 ») a créé le régime dit de la « RTU économique », en ajoutant les dispositions suivantes à l’article L. 5121-12-1 du code de la santé publique : « V. Par dérogation au I et à titre exceptionnel, en présence d'alternative médicamenteuse appropriée disposant d'une autorisation de mise sur le marché, une spécialité pharmaceutique peut faire l'objet d'une recommandation temporaire d'utilisation établie dans les conditions prévues aux I à IV. Cette recommandation temporaire d'utilisation ne peut être établie que dans l'objectif soit de remédier à un risque avéré pour la santé publique, soit d'éviter des dépenses ayant un impact significatif sur les finances de l'assurance maladie » (soulignements ajoutés). Toutefois, le décret d’application n’a jamais été publié. Les représentants de l’autorité nationale de sécurité des médicaments (ci-après, « ANSM »), interrogés par les services d’instruction, ont déclaré en audition que le Conseil d’État avait émis des réserves sur la conformité de l’article L. 5121-12-1 nouveau du code de la santé publique au droit de l’Union, en ce qu’il aurait pu autoriser un « contournement » de l’obligation d’AMM pour des raisons économiques (cote 14274).
27. L’article L. 5121-12-1 du code de la santé publique a donc été de nouveau modifié par la loi n° 2014-892 du 8 août 2014 de financement rectificative de la sécurité sociale pour 2014, comme suit : « I. Une spécialité pharmaceutique peut faire l'objet d'une prescription non conforme à son autorisation de mise sur le marché en l'absence de spécialité de même principe actif, de même dosage et de même forme pharmaceutique disposant d'une autorisation de mise sur le marché ou d'une autorisation temporaire d'utilisation dans l'indication ou les conditions d'utilisation considérées, sous réserve qu'une recommandation temporaire d'utilisation établie par l'Agence nationale de sécurité du médicament et des produits de santé sécurise l'utilisation de cette spécialité dans cette indication ou ces conditions d'utilisation et que le prescripteur juge indispensable le recours à cette spécialité pour améliorer ou stabiliser l'état clinique de son patient » (soulignements ajoutés).
28. Un décret d’application n° 2014-1703, créant les articles R. 5121-76-1 et suivants du code de la santé publique, a ensuite été adopté le 30 décembre 2014 pour préciser les conditions d’application de ces dispositions. Le groupement professionnel « Les entreprises du médicament » (ci-après, « LEEM »), Roche et Novartis ont déposé devant le Conseil d’État un recours pour excès de pouvoir contre ce décret, qui a été rejeté par arrêt du 29 juin 2016 (CE, LEEM, Roche SAS, Novartis Europharm Limited et autres, n° 387890).
b)
Les modalités de vente en ville et à l’hôpital
29. Les modes de fixation des prix des médicaments varient en fonction du canal de vente.
Les modalités de vente en ville
30. La fixation du prix des médicaments s’inscrit dans un cadre juridique complexe, impliquant plusieurs acteurs.
31. Le laboratoire pharmaceutique qui souhaite commercialiser un médicament et le voir pris en charge par l’assurance maladie, doit tout d’abord déposer auprès de la Haute Autorité de santé (ci-après, « HAS ») un dossier, ce qui enclenche une série de procédures : (i) des avis de la HAS portant évaluation du service médical rendu par le médicament (ci-après, « SMR »), de l’amélioration du service médical rendu (ci-après, « ASMR ») par le médicament et de l’efficacité médico-économique du médicament ; (ii) un avis du Comité économique des produits de santé (ci-après, « CEPS »)(7) portant fixation du prix de vente du médicament ; (iii) un arrêté ministériel portant admission au remboursement par l’assurance maladie ; et (iv) la fixation du taux de remboursement par le directeur général de la Caisse nationale d’assurance maladie des travailleurs salariés (ci-après, « CNAMTS ») en sa qualité de directeur de l’Union nationale des caisses d’assurance maladie (ci-après, « UNCAM »).
32. L'article L. 162-16-4 du code de la sécurité sociale, qui détermine les règles de la fixation du prix des médicaments remboursables par la sécurité sociale, précise que « la fixation de ce prix tient compte principalement de l’amélioration du service médical rendu apportée par ce médicament, des prix des médicaments à même visée thérapeutique, des volumes de ventes prévus ou constatés, ainsi que des prévisions prévisibles et réelles d’utilisation du médicament ».
33. Compte tenu du caractère non limitatif des critères énoncés par le code de la sécurité sociale, d’autres critères sont également pris en compte, issus en particulier d’un accord-cadre pluriannuel (trois années) passé depuis 1994 par le CEPS (ou son prédécesseur) avec le LEEM – l’accord actuellement en vigueur étant daté du 31 décembre 2015, prorogé par avenant du 23 juillet 2020 jusqu’au 31 décembre 2020 –, de la « doctrine » formalisée par le CEPS dans son rapport annuel d’activité et des lettres d’orientations ministérielles périodiques.
34. Ainsi, dans un premier temps, le médicament est évalué par la Commission de la transparence de la HAS, qui va rendre un avis portant sur le SMR et sur l’ASMR. La HAS évalue ainsi principalement l’ASMR – qui retrace le progrès thérapeutique relatif apporté par un médicament pour une indication thérapeutique donnée – au regard des médicaments déjà disponibles sur le marché. Selon le cas, son avis indique un progrès thérapeutique majeur (ASMR I), important (ASMR II), modéré (ASMR III), mineur (ASMR IV) ou inexistant (ASMR V).
35. Comme le souligne la HAS, l’évaluation de l’ASMR repose sur une « approche relative », qui présuppose que « 1/ une comparaison soit disponible ; 2/ un comparateur cliniquement ‘pertinent’ ait été identifié ; 3/ et que les données disponibles de la science permettent d’apprécier l’apport du médicament par rapport à ce comparateur. Un comparateur cliniquement ‘pertinent’ (…) se situe au même niveau de la stratégie thérapeutique que le nouveau médicament et est destiné aux mêmes patients. Ainsi, un médicament bénéficiant d’un PTT, d’une autorisation temporaire d’utilisation (ATU), d’une recommandation temporaire d’utilisation (RTU) ou utilisé hors AMM en pratique courante dans l’indication évaluée peut être considéré par la HAS comme un comparateur cliniquement pertinent » (HAS, « Évaluation des médicaments / Doctrine de la commission de la transparence / Principes d’évaluation de la CT relatifs aux médicaments en vue de leur accès au remboursement », septembre 2018). Le CEPS peut ainsi légalement tenir compte de l'innovation apportée par la spécialité au regard des spécialités à même visée thérapeutique précédemment utilisées pour soigner l'affection considérée, en ce compris celles utilisées hors autorisation de mise sur le marché (voir notamment l’arrêt du Conseil d’État du 20 mars 2013, Addmedica, n° 356661 ; voir également le rapport annuel du CEPS pour l’année 2018, page 34).
36. Toutefois, comme l’a rappelé la Cour des comptes dans son rapport sur la sécurité sociale en 2011, la Commission de la transparence de la HAS a « un rôle juridiquement seulement consultatif ». En effet, aucune disposition législative ou réglementaire n’impose que la fixation par le CEPS du prix d’un médicament remboursable ait lieu après avis de cette Commission (voir notamment l’arrêt du Conseil d’État du 20 mars 2013, Addmedica, n° 356661 ; voir également le rapport de la Cour des comptes sur la sécurité sociale 2011, page 124). Le CEPS indiquait en ce sens dans son rapport annuel pour l’année 2010 : « L’ASMR fait l’objet d’un avis de la commission de la transparence. Cet avis est suivi par le comité dans la quasi-totalité des cas. Exceptionnellement toutefois, conformément à la jurisprudence, le comité peut fonder la décision qu’il prend sur une appréciation différente de celle de la commission. Le comité peut considérer sans ASMR un médicament auquel la commission de la transparence en avait reconnu une ou reconnaître une ASMR à un médicament auquel celle-ci n’en avait pas attribué » (page 61). Toutefois, la loi de financement de la sécurité sociale pour 2017 a mis fin à la faculté pour le CEPS, « instance administrative et non scientifique », de se fonder sur une appréciation de l’intérêt thérapeutique plus favorable que celle évaluée par la HAS (Cour des comptes, rapport sur l’application des lois de financement de la sécurité, septembre 2017 ; voir également le rapport annuel du CEPS pour l’année 2016, page 107).
37. Dans un second temps, le prix de vente au public est fixé par convention entre l'entreprise exploitant le médicament et le CEPS. À défaut d’accord, le prix peut être fixé par décision unilatérale du CEPS (article L. 162-16-4 du code de la santé publique).
38. Dans son rapport annuel pour l’année 2008, le CEPS a souligné que : « La discussion du prix d'un médicament avec ASMR constitue donc une négociation ouverte où se confrontent les exigences de l'entreprise et la nécessité ou l’urgence plus ou moins grandes, en termes de satisfaction des besoins de santé, que le médicament soit inscrit au remboursement » (page 54).
39. Pour les médicaments ayant une ASMR I à IV, le prix est fixé par comparaison au prix de « comparateurs », à savoir des médicaments figurant dans la même classe thérapeutique. Ainsi, chaque médicament peut obtenir un avantage tarifaire d’autant plus élevé que son ASMR est importante par rapport aux « comparateurs » identifiés. Pour les médicaments n’apportant pas d’ASMR (ASMR V), le prix doit permettre d’obtenir un coût de traitement inférieur aux « comparateurs » : ils ne peuvent être admis au remboursement par l’assurance maladie qu’à la condition qu’ils apportent « une économie dans le coût de traitement médicamenteux » (article R. 163-5-1-2 du code de la sécurité sociale).
40. Le régime des conventions conclues entre le laboratoire et le CEPS pour fixer le prix de vente public d’un médicament bénéficiant d’une AMM est précisé par l’accord-cadre signé entre le LEEM et le CEPS. L’accord-cadre de 2003 a défini les grandes lignes d’une politique globale du médicament, toujours en vigueur, prévoyant notamment la reconnaissance de l’innovation par l’octroi de prix stables durant cinq ans pour les produits disposant d’une ASMR I, II et III et, sous certaines conditions, d’une ASMR IV (appelée la « garantie de prix européen »)(8). Surtout, pour ces médicaments, la discussion du prix d’inscription à la liste des médicaments remboursables (appelé « prix de liste ») est le plus souvent accompagnée de la discussion de clauses conventionnelles, aussi importantes que le prix lui-même puisqu’elles ont pour objet le plus fréquent d’assurer la meilleure adéquation possible de l’usage du médicament aux besoins identifiés ou de prévoir, en fonction de l’importance des sommes en cause, des rabais de quantité (rapport annuel du CEPS pour l’année 2008). En 2017, la Cour des comptes a souligné qu’« Il n’existe pas d’échelle fixe des avantages tarifaires que peut accepter le CEPS : leur niveau est déterminé lors de chaque négociation » (rapport sur l’application des lois de financement de la sécurité, septembre 2017).
41. Encadrées par la loi (article L. 162-18 du code de la sécurité sociale), les remises correspondent aux sommes dues en exécution de clauses conventionnelles entre le CEPS et les entreprises pharmaceutiques. Il s’agit des sommes versées par le laboratoire à l’assurance maladie, qui viennent en déduction du coût total encouru au titre du remboursement d’un médicament donné pour l’assurance maladie. En 2013, les engagements de type « prix/volume » représentaient 75 % des remises dues, tandis que les remises dites « à la première boîte » représentaient 13 % des sommes dues (rapport annuel du CEPS pour l’année 2013, page 47). En 2017, la part des remises calculées à partir de seuils de volume dans le montant global des remises conventionnelle est restée prépondérante (Cour des comptes, rapport sur l’application des lois de financement de la sécurité, septembre 2017, page 362). Dans son rapport annuel pour l’année 2012, le CEPS a précisé que les remises sont progressivement remplacées par des baisses de prix, lorsque la période de « garantie de prix européen » du médicament concerné est arrivée à échéance (pages 32 et 66).
42. Enfin, les prix fixés par convention peuvent être révisés, à la demande du laboratoire pharmaceutique ou du CEPS. Ce dernier a souligné, dans son rapport annuel portant sur l’année 2012, que « S’agissant des produits bénéficiant d’une garantie de prix européen, le prix et les clauses afférentes peuvent être malgré tout révisés dès lors que survient une modification des conditions qui les avaient justifiés : prix européens, variation importante des coûts de production, évaluation de la spécialité, analyse médico-économique et volumes de ventes constatés » (page 100).
43. Concernant les médicaments « me-too » (c’est-à-dire dont le développement s'est inspiré, avec un niveau de risque assez faible, du succès de leurs prédécesseurs, arrivant donc sur le marché un grand nombre d’années après la mise sur le marché des premiers de la catégorie) commercialisés par une entreprise concurrente, ils ont une ASMR V (prévue pour les médicaments qui n’apportent aucune amélioration du service rendu). Pour fixer leur prix, l’objectif du CEPS est « d’obtenir l’économie la plus importante possible » (rapport annuel du CEPS pour l’année 2017, page 112).
Les modalités de vente à l’hôpital
44. Les prix des médicaments vendus aux hôpitaux sont fixés librement, dans le cadre de procédures d'appels d'offres ou de marchés négociés.
45. Encadrée par le code des marchés publics, la procédure d'achat des hôpitaux publics peut être mise en œuvre par le biais soit d'appels d'offres, garantissant la mise en concurrence des fournisseurs, soit de marchés négociés, sans mise en concurrence, pour les spécialités pharmaceutiques protégées par un brevet ou n’étant confrontées à aucune concurrence (cas limitativement prévus au 8° du II de l'article 35 du code des marchés publics).
46. Par ailleurs, les hôpitaux publics et les cliniques privées peuvent, au choix, passer des marchés individuels ou des marchés collectifs, dans le cadre de groupements de commandes publiques (groupements d'achat), ou encore recourir à des centrales de référencement, qu'ils mandatent pour passer un appel d'offres en leur lieu et place.
47. De manière générale, le financement des médicaments dans les établissements de santé soumis à la tarification à l’activité (appelée « T2A ») est assuré au titre des tarifs des prestations d’hospitalisation afférents aux groupes homogènes de séjour (ci-après, « GHS »). Ces tarifs sont forfaitaires et sont destinés à couvrir l’ensemble des charges supportées par l’établissement pour la prise en charge du patient, et notamment l’ensemble des médicaments administrés durant son séjour hospitalier, qu’il s’agisse d’une hospitalisation complète ou de jour (Rapport IGAS, « Évaluation du dispositif de financement des médicaments en sus des prestations d’hospitalisation dans les établissements de santé », avril 2012, page 16).
48. Par ailleurs, en 2004, un dispositif spécifique de financement des médicaments innovants et onéreux dans les établissements de santé a été mis en place, lors de la mise en œuvre de la tarification à l’activité, afin de garantir à tous les patients un accès à ces médicaments, les tarifs des GHS ne pouvant correctement les prendre en compte. Ce dispositif, mis en place par la loi n° 2003-1199 du 18 décembre 2003 de financement de la sécurité sociale pour 2004, a permis que ces produits soient pris en charge à 100 % par les régimes obligatoires d’assurance maladie s’ils étaient inscrits sur une liste spécifique, appelée la « liste en sus » (article L. 162-22-7 du code de la sécurité sociale).
49. En 2008, à l’époque des pratiques en cause, les médicaments utilisés à l’hôpital étaient financés à plus de 40 % au titre des tarifs des GHS. Leurs prix se forment selon les règles relatives à l’achat public et le CEPS n’intervient donc pas (rapport d’activité du CEPS pour l’année 2008, page 11).
Les liens existant entre la distribution des médicaments à l’hôpital et à la ville
50. Il existe des liens économiques et factuels entre la distribution des médicaments à l’hôpital et en ville.
51. En premier lieu, le secteur pharmaceutique se caractérise par l’existence d’un « effet de réputation », qui correspond à la réputation acquise par le laboratoire qui commercialise le médicament ou la spécialité brevetée, facilitant son référencement par les établissements hospitaliers ou sa prescription par les médecins de ville (décision n° 17-D-25 du 20 décembre 2017 relative à des pratiques mises en œuvre dans le secteur des dispositifs transdermiques de fentanyl, point 53).
52. En deuxième lieu, certains médicaments, prescrits et délivrés initialement à l’hôpital au cours d’une hospitalisation, peuvent être ensuite prescrits et délivrés en ville, quand le patient a regagné son domicile. Il s’agit de « l’effet source », qui désigne la propension des prescriptions délivrées à l’hôpital à générer des ventes en officine de ville, notamment par le biais de renouvellements prescrits par des médecins de ville (voir les décisions n° 10-D-02 du 14 janvier 2010 relative à des pratiques mises en œuvre dans le secteur des héparines à bas poids moléculaire, point 73, et n° 17-D-25 du 20 décembre 2017 relative à des pratiques mises en œuvre dans le secteur des dispositifs transdermiques de fentanyl, point 50).
53. En troisième lieu, certains médicaments peuvent être prescrits à l’hôpital, par exemple lors d’une sortie d’hospitalisation ou au cours d’une consultation externe, mais être délivrés par une officine de ville. Dans ce cas, l’acte de prescription est délivré par le médecin hospitalier, mais le patient achète le médicament dans une pharmacie. Une telle pratique a pu être constatée s’agissant de médicaments « irremplaçables et coûteux », remboursés à 100 % par la sécurité sociale. Tel est le cas, en particulier, d’une des spécialités en cause dans la présente décision : Lucentis (cotes 13595, 13585, 13589 et 7089).
2. LES PRODUITS CONCERNES
54. Après une présentation des anti-VEGF (a), seront présentés les médicaments développés par le laboratoire Genentech (b) et les autres médicaments utilisés dans le traitement de la DMLA (c).
a) Généralités sur les anti-VEGF
Les anti-VEGF
55. L’angiogénèse est le processus de croissance de nouveaux vaisseaux sanguins à partir de vaisseaux préexistants. En soi, elle constitue un processus physiologique normal (notamment lors du développement embryonnaire) mais peut aussi constituer un processus pathologique, par exemple lorsqu’il se traduit par la croissance des tumeurs malignes liées au cancer et le développement des métastases.
56. Les médicaments concernés, dits « anti-VEGF », sont des inhibiteurs de l’angiogénèse, permettant de bloquer la formation des vaisseaux sanguins à partir de vaisseaux préexistants dans le corps humain.
57. Ils sont issus de la découverte en 1989 par le docteur Napoleone Ferrara, biologiste du laboratoire américain Genentech, d’une protéine appelée facteur de croissance de l’endothélium vasculaire (en anglais, « vascular endothelial growth factor », ci-après « VEGF »), qui joue un rôle crucial dans la fabrication des vaisseaux sanguins. Ainsi, en inhibant la production de cette protéine, il est devenu possible de bloquer le phénomène de vascularisation dans le corps. Les chercheurs de Genentech ont alors développé un anticorps permettant l’inhibition du facteur VEGF chez l’humain.
58. À l’origine, les anti-VEGF ont été utilisés pour traiter certains types de cancers, en inhibant le développement vasculaire des tumeurs cancéreuses. Ensuite, les anti-VEGF ont également permis de traiter certains troubles ophtalmologiques liés à une vascularisation excessive de l’œil, comme notamment la dégénérescence maculaire liée à l’âge (ci-après, « DMLA »), par le biais d’injections intravitréennes.
La DMLA et son traitement par les anti-VEGF
59. La DMLA est la principale cause de malvoyance chez les sujets âgés dans les pays industrialisés. À son stade tardif, la DMLA entraîne une altération sévère de la vision centrale, qui se présente notamment sous la forme de tâches sombres perçues par le patient au milieu de sa vision oculaire. Il existe deux formes de DMLA : la forme atrophique (appelée « DMLA sèche ») et la forme néovasculaire ou exsudative (appelée « DMLA humide ou exsudative »). Cette dernière est caractérisée par la croissance anormale de nouveaux vaisseaux sanguins dans la partie centrale de la rétine, la macula.
60. Selon les recommandations de la HAS, les anti-VEGF sont indiqués en première intention pour le traitement de la DMLA exsudative. À ce jour, il n’existe pas de traitement pour la DMLA sèche (cotes 14359 à 14410).
b) Les médicaments développés par le laboratoire Genentech
61. À l’issue de ses recherches sur l’inhibition du VEGF, le laboratoire Genentech a développé deux molécules : le bevacizumab (dont le nom commercial est Avastin) et le ranibizumab (dont le nom commercial est Lucentis).
Avastin
62. À l’origine, le laboratoire Genentech a développé le bevacizumab, en 1996, pour le traitement de certains cancers.
63. Le laboratoire Genentech assure la commercialisation du médicament Avastin aux États-Unis. Pour le reste du monde, le laboratoire a accordé une licence pour l’exploitation et la commercialisation d’Avastin au laboratoire Roche (voir les paragraphes 199 à 209 ci-après).
64. Avastin est un médicament de réserve hospitalière(9), commercialisé en France par le laboratoire Roche, conformément à l’AMM européenne délivrée, le 12 janvier 2005, pour le traitement de certains types de cancers.
65. Toutefois, certains médecins, après avoir administré Avastin en oncologie, ont observé que l’état de santé des patients atteints à la fois d’une tumeur cancéreuse et de DMLA s’améliorait également en ce qui concerne cette dernière pathologie. Ils ont ainsi décidé d’utiliser Avastin en dehors de l’indication prévue dans son AMM, dès 2006, mais également après l’arrivée de Lucentis sur le marché en 2007, en s’appuyant notamment sur des publications scientifiques remontant à 2005 (cotes 7105 et 7106, cote 13584).
66. La pratique consistant à utiliser Avastin pour le traitement de la DMLA s’est alors développée en France, compte tenu notamment de son coût moins élevé que celui de Lucentis (cf. paragraphe 80 ci-après), et ce, même après l’admission de Lucentis au remboursement (cotes 7105 et 7106, cote 7089, cote 7095). En effet, les médecins utilisaient un seul flacon d’Avastin, dont le prix était en 2010 de 348,10 euros (cote 2138) pour effectuer plusieurs injections, le coût unitaire de l’injection étant ainsi ramené à environ 30 euros (cotes 7104 et 2430). Par ailleurs, Avastin était utilisé, toujours « hors AMM », pour d’autres indications ophtalmologiques pour lesquelles Lucentis n’avait pas, ou pas encore, d’AMM (cote 7096, cote 13584).
67. Compte tenu du développement important dans différents pays occidentaux (comme l’Italie, l’Allemagne ou l’Espagne, mais également aux États-Unis) de l’usage d’Avastin pour traiter la DMLA et d’autres affections oculaires, les autorités de santé et équipes médicales ont développé différents projets de recherche visant à tester l’efficacité et les éventuels effets secondaires associés à la prescription d’Avastin pour le traitement de la DMLA.
68. Le laboratoire Roche n’a jamais souhaité demander une AMM pour Avastin dans le domaine ophtalmique (cote 5245, cote 5520).
69. Fin 2011, la création des RTU par la loi Bertrand (cf. paragraphe 22 ci-dessus) a, en pratique, réduit les possibilités pour les médecins ophtalmologistes d’utiliser Avastin pour le traitement de la DMLA, dans la mesure où il existait une spécialité disposant d’une AMM pour cette indication, en l’occurrence Lucentis (cotes 5016 et 5017). Toutefois, interrogée par les services d’instruction, l’ANSM a indiqué, en audition, que « Même après la loi Bertrand, un médecin peut considérer que Lucentis n’était pas approprié pour son patient et choisir Avastin, dans l’intérêt du patient. Le texte n’a pas pour but de verrouiller les choses. Il doit pouvoir coller à la pratique médicale. Cela doit être fondé sur des éléments scientifiques sérieux. Ce n’est pas un interdit absolu. Cela doit respecter des critères : intérêt du patient et transparence » (cote 14274).
70. Au cours de l’année 2012, les pouvoirs publics ont commencé à évoquer la possibilité de mettre en œuvre une RTU pour Avastin. Ainsi, à la fin du mois de juin 2012, M. X…, alors président de la HAS, a annoncé publiquement envisager de saisir l’ANSM en vue d’une RTU pour Avastin dans la DMLA (cote 5852).
71. Néanmoins, dans un courrier du 31 juillet 2012 adressé à Novartis, M. X… a indiqué que le collège de la HAS avait finalement renoncé à cette demande, en se référant aux doutes sur la possibilité juridique d’adopter une RTU en dehors du cas d’absence d’une alternative thérapeutique régulièrement autorisée (« Nous avons finalement renoncé à prendre cette initiative compte tenu du libellé de l’article L. 5121-12-1 qui définit les critères d’élaboration d’une RTU. Le décret du 9 mai 2012 précise en effet qu’une RTU peut être élaborée dans une indication différente ou dans des conditions d’utilisation non conformes à l’AMM en l’absence d’alternative médicamenteuse appropriée autorisée. Bien que le terme ‘appropriée’ puisse faire l’objet de discussions, nous n’avons pas souhaité utiliser la possibilité qui est offerte à la HAS de saisir l’ANSM, notamment dans l’attente des résultats de l’étude GEFAL », cote 3336).
72. Aujourd’hui, l’utilisation d’Avastin est encadrée par une RTU, publiée le 25 juin 2015 et entrée en vigueur le 1er septembre 2015(10). En raison du refus de Roche d’assurer le suivi des patients, la RTU initiale a été modifiée le 11 septembre 2015 afin de permettre aux Hospices civils de Lyon d’assurer cette fonction de suivi des patients(11). Roche et Novartis ont déposé devant le Conseil d’État un recours pour excès de pouvoir contre la décision du directeur général de l’ANSM établissant la RTU pour Avastin, qui a été rejeté par arrêt du 24 février 2017 (CE, Sect. 1 et 6 réunies, 24 février 2017, n° 392459, inédit). La RTU a été renouvelée pour 3 ans le 1er septembre 2018. Le prix de la seringue d’Avastin dans l’indication de sa RTU a été fixé par l’arrêté du 19 août 2015 à un montant de 10 euros. Par un arrêté du 4 mai 2016, ce prix a été modifié et fixé rétroactivement à compter du 1er mars 2016, à un montant de 100 euros TTC(12). Cette spécialité est financée selon le principe de la « liste en sus », c'est-à-dire donnant lieu à un versement pécuniaire à l’hôpital, des prestations d’hospitalisation remboursées par le biais des GHS, conformément aux dispositions de l’article L. 162-17-2-1 du code de la sécurité sociale.
73. Depuis sa publication, la RTU a effectivement permis à certains médecins d’utiliser à nouveau Avastin dans le traitement de la DMLA. À titre d’illustration, le chef du service de pharmacie clinique du groupe hospitalier Hôpitaux universitaires Paris Centre a indiqué sur ce point : « nous avons cessé toute préparation d’Avastin quand nous avons eu connaissance de la circulaire de la DGS de juillet 2012. Je n’ai repris cette production qu’à compter du 1er septembre 2015 suite à la mise en application de la RTU du 24 juin 2015 » (cote 7095 ; voir également, cote 13589).
Lucentis
74. Le laboratoire Genentech a développé, au milieu des années 1990, un fragment d’anticorps destiné spécifiquement à l’utilisation en injection dans l’œil (cote 47559). Selon Genentech, le ranibizumab est indiqué pour un usage dans l’œil en raison de son faible poids moléculaire, ainsi que de l’absence de fragment dit « Fc », cette absence limitant la possibilité d’un passage systémique – c’est-à-dire dans le système sanguin – de l’anticorps.
75. Ainsi, si le bevacizumab (Avastin) et le ranibizumab (Lucentis) ont la même cible moléculaire, à savoir les VEGF, ils n’ont pas la même forme ni le même poids moléculaire. Le bevacizumab (Avastin) est un anticorps monoclonal entier, tandis que le ranibizumab (Lucentis) est un fragment d’anticorps, dont le poids moléculaire est nettement plus faible :

76. Le laboratoire Genentech assure la commercialisation du médicament Lucentis aux États-Unis. Pour le reste du monde, il a accordé une licence pour l’exploitation et la commercialisation de Lucentis au laboratoire Novartis (cf. paragraphes 210 à 220 ci-après).
77. Ainsi, Lucentis est commercialisé en France par le laboratoire Novartis, conformément à l’AMM européenne accordée, le 22 janvier 2007, pour le traitement de la DMLA.
78. Ses indications ont ensuite été étendues au traitement d’autres pathologies oculaires, par AMM européenne accordée le 6 janvier 2011, pour le traitement de l’oedème maculaire diabétique (ci-après, « OMD »), puis par AMM européenne accordée le 27 mai 2011, pour le traitement des occlusions veineuses rétiniennes (plus spécifiquement, occlusion d’une branche veineuse rétinienne, ci-après, « OBVR » et occlusion de la veine centrale de la rétine, ci-après « OVCR »), et de la baisse visuelle due à une néovascularisation choroïdienne (ci-après, « NVC ») secondaire à une myopie forte.
79. Lucentis est remboursé à 100 % en France, pour le traitement de la DMLA (cotes 15188 à 15194). Les autres indications ont été admises au remboursement en 2012. Dans un document relatif aux dépenses des médicaments de ville en 2012, l’Assurance maladie précise que Lucentis est devenu le premier médicament de ville remboursé par la sécurité sociale en France, avec près de 390 millions d’euros remboursés et une progression très forte d’environ 30 % par rapport à 2011 (cote 14200).
80. Au moment de son lancement, en 2007, le prix de Lucentis était de 1 161 euros par dose injectée. Il a ensuite fait l’objet de plusieurs baisses, entre 2008 et 2013, pour atteindre 789,50 euros par dose (cote 45091). Selon le CEPS, le prix a été renégocié à la fin de garantie de prix européenne, en 2012, dans le cadre d’une seule négociation incluant les extensions d’indications (cote 15186).
81. Plus précisément, dans un avis du 28 mars 2007, la Commission de la transparence de la HAS a reconnu à Lucentis un niveau d’ASMR important (niveau II), justifiant que cette spécialité bénéficie de la « garantie de prix européen » pendant 5 ans. Dans un avis du 21 novembre 2012, rendu à l’occasion du renouvellement de l’inscription de Lucentis, dont les indications ont été étendues (cf. paragraphe 78 ci-dessus), le niveau d’ASMR a été maintenu. À ces deux occasions, Avastin n’a pas été retenu comme médicament « comparateur » au titre de l’évaluation de Lucentis. Le CEPS a indiqué en audition sur ce point qu’ « il n’y avait pas de comparateur donc il y a eu un accord prix/volume pour maîtriser l’impact budgétaire. S’il n’y avait pas de remise à la première boite, c’est qu’il n’y avait pas de comparateur » (cote 15185).
82. Ce n’est que postérieurement à l’adoption de la RTU Avastin (cf. paragraphe 72 ci-dessus) que la spécialité Avastin a été retenue comme comparateur de Lucentis. Dans son avis du 11 octobre 2017, intitulé « Place dans la stratégie thérapeutique de Lucentis, Eylea et de leurs comparateurs cliniquement pertinents dans la forme néovasculaire (humide) de la dégénérescence maculaire liée à l’âge (DMLA) », la Commission de la transparence de la HAS a conclu que : « dans le traitement de la forme néovasculaire (humide) de la dégénérescence maculaire liée à l’âge, le comparateur cliniquement pertinent de Lucentis (ranibizumab) et Eylea (aflibercept) est Avastin (bevacizumab), dans le cadre d’une RTU »(13).
c) Les autres médicaments utilisés dans le traitement de la DMLA
83. L’industrie pharmaceutique a développé d’autres médicaments permettant le traitement des maladies oculaires, comme Eylea, Macugen ou encore Visudyne.
Eylea
84. La molécule aflibercept a été développée par le laboratoire américain Regeneron. Cette même molécule a donné lieu au développement de deux produits distincts : d’une part, Zaltrap, indiqué pour certains types de cancers, et commercialisé en Europe par le laboratoire Sanofi et, d’autre part, Eylea, indiqué en injection oculaire intravitréenne, et commercialisé en Europe par le laboratoire Bayer.
85. Eylea, qui est un médicament ayant pour substance active l’aflibercept est également classé comme anti-VEGF (cote 14378), même si son mode d’action est différent de celui du ranibizumab et du bevacizumab (cote 6210).
86. Eylea a fait l’objet d’une AMM européenne le 22 novembre 2012, pour le traitement de la DMLA. Il est commercialisé en France depuis le début du mois de novembre 2013. Son prix, de 710,55 euros HT par dose, a été arrêté par un avis publié au Journal officiel le 30 octobre 2013(14). Ses indications ont ensuite été étendues au traitement d’autres pathologies oculaires : en 2014 pour le traitement de l’OVCR ; en 2015, pour le traitement de l’OMD ; en 2016, pour le traitement de l’OBVR et de la baisse visuelle due à une NVC(15).
Macugen
87. Le pegaptanib (dont le nom commercial est Macugen) est le premier médicament à activité anti-angiogénique, indiqué dans le traitement de la DMLA (cote 14375).
88. Macugen, distribué par le laboratoire Pfizer, dispose d’une AMM pour le traitement de la forme néovasculaire de la DMLA depuis 2006.
Visudyne
89. La verteporfine (dont le nom commercial est Visudyne) a été le premier traitement disponible pour traiter les lésions rétrofovéolaires de la DMLA, appelé traitement par photothérapie dynamique (cote 14379, cote 3804).
90. Visudyne, commercialisé par le laboratoire Novartis, dispose d’une AMM européenne depuis le 27 juillet 2000.
91. Aujourd’hui, le traitement par Visudyne, seul ou en combinaison avec un anti-VEGF, est désormais considéré par la HAS comme devant être appliqué uniquement dans les cas d’absence d’efficacité du traitement par anti-VEGF seul (cotes 14379 et 14380).
3. LE CADRE SCIENTIFIQUE CONCERNANT L’UTILISATION D’AVASTIN DANS LE TRAITEMENT DES PATHOLOGIES OCULAIRES
92. L’efficacité et la sécurité comparée d’Avastin et de Lucentis en ophtalmologie ont fait l’objet de diverses études scientifiques successives (a), qui ont permis aux autorités de santé de prendre position sur l’utilisation d’Avastin pour le traitement de la DMLA et d’autres pathologies oculaires (b).
93. Un grand nombre d’études scientifiques a été consacré au sujet, en raison tant de l’enjeu de santé publique – la DMLA étant l’une des affections oculaires les plus répandues et les plus graves –, que de l’enjeu financier majeur pour les systèmes de santé, les traitements disponibles étant particulièrement onéreux.
a) Les études scientifiques sur l’efficacité et la sécurité comparée d’Avastin et de Lucentis en ophtalmologie
94. Jusqu’en 2010, il existait peu de données scientifiques disponibles concernant la sécurité et l’efficacité d’Avastin en ophtalmologie. Puis, entre 2010 et 2013, plusieurs études médicales, publiées dans plusieurs pays occidentaux, ont fourni des éléments permettant de comparer l’efficacité et la tolérance respective d’Avastin et de Lucentis en usage dans l’œil.
95. Trois catégories d’études ont été menées : des études rétrospectives (c'est-à-dire fondées sur les dossiers médicaux des patients, répertoriant les événements survenus avant la mise en place de l’étude, utilisant la base de données Medicare aux États-Unis), des études prospectives de comparaison, également appelées « Head to Head » ou « H2H » (c'est-à-dire qui intègrent des patients en début ou en cours de traitement et répertorient les évènements survenus après la mise en place de l’étude) et des méta-analyses (qui regroupent et exploitent statistiquement les données de plusieurs études similaires, réalisées sur la base des études cliniques précitées).
96. Comme indiqué par les représentants de l’ANSM, interrogés par les services d’instruction, en 2018, en audition : « Sur Avastin, le débat scientifique a été évolutif. Ce n’est que progressivement que les méta-analyses se sont stabilisées en faveur d’Avastin. Les connaissances ont évolué. Tout cela est contemporain. Les méta-analyses sont arrivées au moment où on étudiait la RTU. Nous avons utilisé cinq grandes études randomisées (CATT, IVAN, GEFAL, MANTA, LUCAS) et deux méta-analyses, Solomon (plutôt orientée efficacité) et Moja (qui a apporté des éléments de sécurité déterminants) » (cote 14274).
Les études rétrospectives
97. Dans un premier temps, deux études rétrospectives, utilisant la base de données Medicare aux États-Unis, ont été réalisées : l’étude Curtis et al. et l’étude Gower et al.
L’étude Curtis et al. (résultats publiés en octobre 2010)
98. L’objectif de l’étude Curtis et al., publiée en octobre 2010, était de comparer le profil de sécurité des molécules de ranibizumab (Lucentis) et de bevacizumab (Avastin) avec les traitements utilisés antérieurement à base de photothérapie ou utilisant la molécule de pegaptanib (Macugen) (cotes 14525 à 14531).
99. La conclusion principale de cette étude est que tant Lucentis qu’Avastin ne présentent pas de risque accru d’effets secondaires graves par rapport aux traitements antérieurs. Toutefois, l’étude note qu’une comparaison entre Lucentis et Avastin montre un risque plus faible de mortalité et d’accident vasculaire cérébral pour les patients traités par Lucentis, que pour ceux traités par Avastin.
100. Afin de vérifier que cette différence n’était pas liée aux caractéristiques socio-économiques de la population étudiée (les patients traités avec Lucentis pouvant être issus d’un milieu socio-professionnel plus aisé, compte tenu du prix du traitement), les auteurs ont analysé un échantillon plus réduit de patients traités dans des centres fournissant exclusivement soit Lucentis, soit Avastin. Pour cet échantillon, aucune différence pertinente statistiquement entre les deux traitements n’a été relevée (« There were no significant differences in study outcomes between the treatment groups », cote 14529).
L’étude Gower et al. (résultats rendus publics en avril 2011)
101. L’étude Gower et al., financée par Genentech, portait sur l’analyse rétrospective des effets indésirables après une injection d’Avastin ou de Lucentis dans le traitement de la DMLA. Elle n’a pas fait l’objet d’une publication dans son intégralité dans une revue médicale, seul un résumé ayant été publié en avril 2011 (cotes 14532 et 14533 ; cotes 5612 à 5616 ; cote 16737).
102. La conclusion de cette étude est, selon le résumé disponible, que les données issues du Medicare suggèrent des différences entre Lucentis et Avastin quant à leur profil de tolérance. Toutefois, les auteurs reconnaissent que cette étude présente des limites compte tenu des informations incomplètes quant aux facteurs de comorbidité(16) (« Data from this Medicare claims analysis suggest differences in the safety profile of Bev [Avastin] vs Ran [Lucentis]. However, this study is limited by incomplete information on some important confounding factors, e.g. smoking, lipid and blood pressure levels, which would further clarify the relative safety of these treatments in wet AMD », cote 14533).
103. Plus précisément, le résumé fait ressortir, concernant les effets systémiques(17), un risque de mortalité de 11 % supérieur et un risque d’AVC hémorragique supérieur de 57 % avec Avastin. Concernant les effets oculaires, il relève un risque d’inflammation oculaire supérieur de 80 % avec Avastin. Toutefois, le résumé précise que ces différences ont été atténuées dans le cadre d’une analyse secondaire, portant sur une période plus longue (« Differences in overall mortality and hemorrhagic CVA were attenuated in secondary analyses that included use of Bev [Avastin] or Ran [Lucentis] on unclassified drug codes and data back to 2006 », cote 14533).
Les études de comparaison
104. Plusieurs essais cliniques indépendants, publiés entre 2011 et 2014, ont comparé l’efficacité et la sécurité d’Avastin et de Lucentis dans le traitement de la DMLA. Parmi ceux-ci, les études CATT, IVAN et GEFAL ont tenu une place particulièrement importante dans le débat public.
L’étude CATT (résultats à un an publiés en mai 2011 et à deux ans en juillet 2012)
105. L’étude CATT était un essai multicentrique (44 sites) dit de « non-infériorité » réalisé aux États-Unis entre février 2008 et décembre 2009. Son objectif principal était de démontrer la non-infériorité d’Avastin par rapport à Lucentis, en termes d’efficacité clinique à un et deux ans sur l’acuité visuelle de patients atteints de DMLA.
106. Les résultats de l’étude à un an ont été publiés en mai 2011 (cotes 53 à 78), et ceux de l’étude à deux ans, en juillet 2012 (cotes 14514 à 14524).
107. À un an, la conclusion de l’étude est que le bevacizumab (Avastin) et le ranibizumab (Lucentis) ont des effets équivalents sur l’acuité visuelle des patients traités selon le même protocole. En d’autres termes, les auteurs de l’étude ont conclu à la non-infériorité d’Avastin par rapport à Lucentis en termes d’efficacité clinique sur l’acuité visuelle des patients.
108. Concernant les données de tolérance, observée en tant que critère secondaire dans l’étude, l’étude CATT indique n’avoir trouvé aucune différence significative entre les deux molécules en termes de mortalité, d’événements artériothrombotiques ou d’événements thrombotiques veineux. Toutefois, le taux général d’événements indésirables systémiques était plus important pour les patients traités avec Avastin que ceux traités avec Lucentis (24,1 % contre 19 %). À cet égard, les auteurs notent que ces effets indésirables concernaient des affections diverses, qui pour la plupart n’avaient pas été identifiées lors des tests pour l’utilisation d’Avastin contre le cancer. Ils concluent donc que des études supplémentaires sont nécessaires pour déterminer la cause de ces différences en termes de tolérance (« Différences in rates of serious adverse events require further study », cote 53). De manière générale, les auteurs notent que la puissance statistique de leur étude est limitée, en ce qui concerne la détection de certains événements indésirables importants (« With a limited statistical power to detect important advese events, we found no significant differences between the two drugs in rates of death, arteriothrombotic events, or venous thrombotic events », cote 63).
109. À deux ans, la conclusion générale de l’étude sur l’efficacité comparée du bevacizumab (Avastin) et du ranibizumab (Lucentis) est identique : les deux spécialités ont des effets équivalents sur l’acuité visuelle des patients.
110. En termes de tolérance, la conclusion générale est qu’il n’y a pas de différence entre les deux molécules concernant les taux de mortalité et d’événements artériothromboliques (« Rates of death and arteriothrombotic events were similar for both drugs », cote 15514). En outre, les auteurs estiment que l’interprétation de la persistance d’un taux plus élevé d’événements indésirables pour Avastin (31,7 % des patients traités par Lucentis, contre 39,9 % des patients traités par Avastin) est incertaine, en raison du fait que ces effets ne peuvent pas être reliés à des affections généralement associées à l’inhibition des VEGF (« The interpretation of the persistence of higher rates of serious adverse events with bevacizumab [Avastin] is uncertain because of the lack of specificity to conditions associated with inhibition of VEGF », cote 14514).
L’étude IVAN (résultats à un an publiés en 2012 et à deux ans en juillet 2013)
111. L’étude IVAN est un essai multicentrique de non-infériorité réalisé en Grande-Bretagne entre mars 2008 et octobre 2010. Son objectif principal était de comparer, à deux ans, l’efficacité et la sécurité du ranibizumab (Lucentis) et du bevacizumab (Avastin) en injections intravitréennes pour le traitement de la DMLA.
112. Les résultats de l’étude à un an ont été publiés en juillet 2012 (cotes 66 à 78), ceux à deux ans, en juillet 2013 (cotes 47135 à 47145). Les auteurs précisent que les résultats primaires étant à deux ans, les résultats présentés à un an sont provisoires.
113. Dans leur article présentant les résultats à un an, les auteurs constatent que le bevacizumab (Avastin) n’est ni inférieur, ni équivalent au ranibizumab (Lucentis), et que, partant, la comparaison des spécialités est non-conclusive (« The comparison is inconclusive ; bevacizumab [Avastin] was neither inferior nor equivalent to ranibizumab [Lucentis] using the 3,5 letter limit », cote 73). Toutefois, ils soulignent que la moyenne des différences entre les deux molécules est de deux lettres en faveur du rabinizumab (Lucentis), précisant néanmoins qu’il s’agit d’une faible différence d’un point de vue clinique (« The mean difference between the drugs was 2 letters in favor of ranibizumab [Lucentis], a small difference from a clinical perspective », cote 74).
114. Concernant les données de tolérance, les auteurs notent que le profil de sécurité des deux molécules est rassurant (« The safety profiles of the drugs were reassuring », cote 75). S’agissant des événements artériothrombotiques ou des problèmes cardiaques, qui constituaient un critère de sécurité principal pour l’étude, ces effets indésirables sont restés rares (moins de 2 %) mais se sont produits plus fréquemment avec le ranibizumab (Lucentis) qu’avec le bevacizumab (Avastin). Enfin, les auteurs constatent qu’il n’existait pas de différence entre les deux molécules dans les pourcentages subissant des événements indésirables systémiques (« There was no difference between drugs in the proportion experiencing a serious systemic adverse event », cote 66). S’agissant du passage dans le sang du produit, après avoir constaté que le bevacizumab (Avastin) réduit les niveaux systémiques de VEGF dans une plus grande mesure que le ranibizumab (Lucentis), l’étude indique qu’il est possible que les conséquences de cette différence n’apparaissent qu’à l’issue d’un plus long suivi (« it is possible that consequences of differential suppression of circulating VEGF will only become apparent after longer follow-up », cote 75).
115. Prenant également en compte les résultats des autres études disponibles (en particulier, l’étude CATT susvisée), les auteurs concluent que le ranibizumab (Lucentis) et le bevacizumab (Avastin) ont des bénéfices équivalents en termes de fonctions visuelles. Par ailleurs, ils notent que les deux spécialités ont des profils de sécurité similaires et ne démontrent pas un risque accru d’événements artériothrombotiques avec le bevacizumab (Avastin). En revanche, ils soulignent que les études CATT et IVAN ont toutes deux démontré une faible proportion supérieure d’événements indésirables systémiques avec le bevacizumab (Avastin).
116. À deux ans, les auteurs constatent à nouveau que le critère de la non-infériorité en termes d’acuité visuelle entre les deux molécules est non conclusif, pour les mêmes raisons que lors de l’analyse à un an. Ils notent que la différence moyenne constatée entre les groupes, en faveur du ranibizumab (Lucentis) est faible, soulignant que selon les critères, moins stricts de l’étude CATT (différence de cinq lettres, au lieu de 3,5 lettres(18) pour la présente étude), la non-infériorité aurait été établie (« Non-inferiority for both comparisons would have been established had we used the CATT non-inferiority margin of 5 letters », cote 47142).
117. Concernant les données de tolérance et de sécurité, les auteurs indiquent préférer avoir recours, pour des raisons de puissance statistique, à une analyse combinée des études CATT et IVAN. Ils confirment ainsi qu’à deux ans, la comparaison entre les deux molécules est rassurante, soulignant qu’aucune différence n’a été identifiée, en termes de mortalité ou d’événements artériothrombotiques, ceux-ci pouvant être liés à l’utilisation d’anti-VEGF. Ils relèvent que l’analyse combinée des événements systémiques indésirables sévères semble confirmer un risque augmenté pour le bevacizumab (Avastin), comme cela a été identifié dans l’étude CATT, tout en soulignant que la combinaison des résultats des deux études supprime les différences constatées entre les estimations des différentes études (« The pooled analysis for any systemic SAE [systemic adverse events] seems to confirm an increased risk with bevacizumab [Avastin], which was first reported in CATT. However, the pooled analysis disguises the inconsistency between the separate trial estimates », cote 47143).
118. En conclusion, les auteurs constatent, au regard de l’étude IVAN et des méta-analyses des données CATT et IVAN, que le ranibizumab (Lucentis) et le bevacizumab (Avastin) ont une efficacité similaire et peuvent être considérés comme équivalents pour le traitement de la DMLA.
L’étude GEFAL (résultats rendus publics en mai 2013)
119. L’étude GEFAL est une étude multicentrique (38 sites) de non-infériorité, réalisée en France entre mars 2009 et juillet 2012 par les Hospices civils de Lyon. Elle a été financée par le Ministère de la santé et par la Caisse nationale d’assurance maladie dans le cadre du programme hospitalier de recherche clinique pour 2008. Son objectif principal était de « montrer la non-infériorité, en terme d’efficacité clinique à 12 mois, du bevacizumab [Avastin] par rapport au ranibizumab [Lucentis] sur l’acuité visuelle de patients atteints de dégénérescence maculaire liée à l’âge (DMLA) néovasculaire rétrofovéolaire ». L’étude intégrait également un ensemble d’objectifs secondaires, notamment : « évaluer et comparer la tolérance à 12 mois des traitements par bevacizumab [Avastin] et ranibizumab [Lucentis] au niveau local et systémique » (cote 2282).
120. Ses résultats ont été publiés en novembre 2013 (cotes 14547 à 14556), mais rendus publics par le Professeur Y…, directeur de l’étude, dès le mois de mai 2013, lors du congrès de l’ARVO (« Association for Research in Vision and Ophthalmology ») (cotes 15846 à 15877).
121. Dans l’article présentant les résultats à un an, les auteurs constatent que le bevacizumab (Avastin) est non-inférieur au ranibizumab (Lucentis).
122. En termes de tolérance, les auteurs soulignent que les profils respectifs de sécurité des deux spécialités semblent similaires. Ils notent qu’aucune différence en termes de mortalité, d’événement artériothrombotiques ou d’événements thrombotiques veineux n’a été identifiée. La proportion des patients subissant des événements indésirables graves est de 12,6 % pour le bevacizumab (Avastin) et de 12,1 % pour le ranibizumab (Lucentis). Les proportions des patients subissant des événements systémiques ou oculaires graves étaient similaires pour les deux spécialités.
123. En conclusion, les auteurs de l’article indiquent que l’étude GEFAL est la deuxième étude à démontrer la similarité d’Avastin et de Lucentis en termes d’efficacité et qu’aucune différence en termes de sécurité n’a pu être identifiée. Toutefois, ils rappellent que cette étude, comme les autres études de comparaison, n’a pas la puissance statistique nécessaire pour identifier avec certitude des différences en termes de sécurité.
124. Lors d’une présentation au congrès ARVO, en mai 2013, le Professeur Y… a également exposé les résultats de la méta-analyse, incluant CATT, IVAN et GEFAL (cotes 15846 à 15877). Il a souligné à cette occasion que les résultats de l’étude GEFAL concordaient avec les études précédentes : s’ils renforcent les constats initiaux de non-infériorité sur l’acuité visuelle et d’une proportion d’événements indésirables systémiques graves plus importante avec le bevacizumab (Avastin), aucune différence n’est constatée en termes de mortalité et d’événement artériothrombotiques, et une méta-analyse individuelle est donc nécessaire avant d’en tirer des conclusions. À ce sujet, le Professeur Y… a indiqué en audition : « Dans la méta-analyse, on a inclu GATT, IVAN, MANTA et GEFAL. Ce n’étaient que des résultats à un an. Nous avons trouvé plus d’effets indésirables graves systémiques avec l’Avastin. Le problème, c’est qu’on n’a pas su identifier pourquoi. Pas de différence sur la mort, sur les APTC (là où on attend la molécule) ou sur les effets gastro-intestinaux, pas de différence non plus » (cote 16623).
Les méta-analyses
125. Par la suite, les études de comparaison présentées ci-dessus ont été combinées dans le cadre de deux méta-analyses publiées en 2014. Celles-ci ont été prises en compte par l’ANSM dans le cadre du projet de RTU pour Avastin(19).
126. Ces méta-analyses étaient fondées sur la méthode dite « Cochrane », par référence à l’organisme éponyme, dont l’objectif est de développer les pratiques de revue systématique de la littérature scientifique dans le domaine médical. En reprenant et analysant toutes les publications disponibles sur une intervention ou un traitement donné, il est ainsi possible de corriger les biais et les risques d’erreurs aléatoires des études individuelles.
127. La première étude – l’étude « Solomon » – combinait efficacité et sécurité et incluait 12 études, dont certaines n’étaient pas des études de comparaison entre Avastin et Lucentis. Concernant les données de sécurité, les résultats font état d’un risque plus important de présenter un événement indésirable grave systémique sous Avastin, sans pouvoir identifier un événement particulier.
128. La seconde étude – l’étude « Moja » – avait comme critère primaire la sécurité des patients, et cherchait à évaluer le nombre et la gravité des effets indésirables constatés avec chaque traitement. Les résultats de l’étude ne dégagent pas d’augmentation significative du risque de présenter un événement indésirable grave systémique avec Avastin par rapport à Lucentis, malgré la plus grande puissance statistique. Cependant, elle observe une augmentation des effets de type gastro-intestinaux pour Avastin, sans pouvoir identifier un effet précis. Cette augmentation confirme la tendance observée dans l’étude « Solomon ».
b) Les prises de position des autorités de santé sur l’efficacité et la sécurité comparée d’Avastin et de Lucentis en ophtalmologie
129. Les études présentées supra ont été utilisées et commentées dans le cadre du débat public sur la possibilité d’avoir recours à Avastin de façon sécurisée en lieu et place de Lucentis.
Les autorités de santé françaises et européennes ont ainsi été amenées à se prononcer sur cette question.
L’analyse des autorités de santé européennes
130. L’Agence européenne du médicament (« European Medicine Agency », ci-après, « EMA »), est chargée de garantir l’évaluation scientifique, le contrôle et le suivi de la sécurité des médicaments à usage humain dans l’Union européenne. Elle a pour mission principale d’autoriser et de contrôler les médicaments dans l’Union européenne. Entre 2011 et 2013, l’EMA a mis en œuvre deux procédures distinctes de modification du résumé des caractéristiques du produit (ci-après, « RCP »)(20), respectivement pour Avastin et pour Lucentis, afin d’y intégrer des informations sur les effets indésirables liés à l’utilisation de ces produits en ophtalmologie.
131. À ces deux occasions, le comité des médicaments à usage humain (ci-après, « CHMP »), l’un des sept comités scientifiques que compte l’EMA, a étudié de manière approfondie les résultats des études scientifiques disponibles sur l’efficacité et la sécurité d’Avastin et de Lucentis en ophtalmologie.
Le changement de RCP d’Avastin
132. Le 20 juin 2011, Roche a demandé la modification du RCP d’Avastin, en vue d’y intégrer des informations portant sur des cas d’inflammations de l’œil et d’endophtalmies intervenus après l’utilisation d’Avastin en injection intravitréenne. Roche demandait également à intégrer, dans la section 4.8 « Effets indésirables », les effets indésirables systémiques identifiés dans le cadre des études rétrospectives et de comparaison avec Lucentis.
133. Le 19 juillet 2012, le CHMP a rendu son avis (cotes 752 à 768).
134. Dans ce cadre, le CHMP a analysé l’ensemble de la littérature scientifique fournie par Roche, en particulier les études Curtis et Gower, les résultats de l’étude CATT à un et deux ans, ainsi que ceux de l’étude IVAN à un an.
135. S’agissant en premier lieu des affections liées à l’œil, le CHMP a considéré qu’il convenait de procéder à une mise à jour de la section 4.4 « Mises en gardes spéciales et précautions d’emploi », afin d’indiquer que des cas d’effets indésirables oculaires ont été reportés après l’utilisation non autorisée d’Avastin en injection intravitréenne.
136. S’agissant en second lieu des effets systémiques, le CHMP a constaté : « detailed safety information provided from the CATT and IVAN studies is reassuring and no evidence can be provided that bevacizumab [Avastin] is systemically more unsafe than ranibizumab [Avastin] and vice-versa » (traduction libre : « les informations de sécurité détaillées obtenues des études CATT et IVAN sont rassurantes et il n’y a pas de preuve que le bevacizumab [Avastin] présente plus de risques d’un point de vue systémique que le ranibizumab [Lucentis] et vice-versa »).
137. Il a toutefois relevé que « a reduction of circulating VEGF concentration has been demonstrated following intravitreal anti-VEGF therapy. In addition, systemic adverse events including non-ocular haemorrhages and ATE [arterial thromboembolic events] events have been reported following to ITV injections of VEGF inhibitors and there is a theoretical risk that these may relate to VEGF inhibition » (traduction libre : « une réduction de la concentration en VEGF circulant dans le sang a été constatée après un traitement par injection intravitréenne d’anti-VEGF. De plus, des effets systémiques indésirables, comprenant des hémorragies ou des événements arthériothrombotiques ont été rapportés après des injections intravitréennes d’inhibiteurs de VEGF et il y a un risque théorique que ceux-ci soient liés à l’inhibition des VEGF »). Ce constat porte ainsi sur l’ensemble des traitements par anti-VEGF, et pas uniquement Avastin.
138. En conclusion, le CHMP a estimé qu’il n’était pas nécessaire de modifier la section 4.8 « Effets indésirables », mais qu’il convenait de modifier la section 4.4 « Mises en gardes spéciales et précautions d’emploi » par une mise à jour de la partie portant sur les affections liées à l’œil, d’une part, et par l’insertion d’un paragraphe sur l’existence d’effets systémiques indésirables liés à l’utilisation des anti-VEGF en intravitréen, d’autre part.
139. La Commission européenne a modifié sa décision d’AMM en ce sens le 30 août 2012.
Le changement de RCP de Lucentis
140. Le 8 février 2012, Novartis a demandé la modification du RCP de Lucentis, en vue d’y intégrer des informations portant sur des risques d’attaques cardiaques et d’infarctus du myocarde liés à l’utilisation de la classe de produit des anti-VEGF, dans la section 4.8 « Effets indésirables ».
141. Cette demande de modification a été réalisée à la demande du CHMP, après analyse du rapport périodique de sécurité de Lucentis, sur la période allant de juillet 2010 à juin 2011, faisant état d’un certain nombre de cas d’infarctus du myocarde et d’événements artériels thromboemboliques relevés chez des patients traités au ranibizumab (Lucentis).
142. Le 17 janvier 2013, le CHMP a rendu son avis (cotes 769 à 796).
143. En introduction, le CHMP a rappelé les constats exposés plus haut (cf. paragraphe 137 de la présente décision) sur l’existence d’un risque théorique que certains effets indésirables systémiques soient liés à la réduction de la concentration en VEGF dans le sang, après un traitement par anti-VEGF : « Following intravitreal injection of ranibizumab, there is a theorical risk of systemic arterial thromboembolic events due to systemic inhibition of VEGF » (traduction libre : « Après une injection de ranibizumab [Lucentis], il y a un risque théorique d’événements systémiques thromboemboliques artériels causé par l’inhibition systémique de VEGF »). Il a souligné que ces effets étaient commun à l’ensemble de la classe des inhibiteurs du VEGF : « this is considered a class effect of anti-VEGF drugs » (traduction libre : « ceci est considéré comme un effet de la classe des médicaments anti-VEGF »).
144. Le CHMP a ensuite procédé à une analyse de l’ensemble de la littérature scientifique fournie par Novartis, incluant les études conduites spécifiquement par le laboratoire et celles comparant Lucentis à d’autres anti-VEGF (Avastin et Eylea), études qui avaient déjà été analysées dans le cadre du changement de RCP d’Avastin.
145. En premier lieu, le CHMP a indiqué que les éléments fournis par Novartis sur les différences pharmacocinétiques existant entre Lucentis et les autres anti-VEGF ne permettaient pas de formuler des recommandations différenciées donnant l’impression que Lucentis serait plus sûr que d’autres anti-VEGF : « the PK and PD data alone were insufficient to justify a differential warning that gives the impression that Lucentis is safer compared to other anti-VEGF treatments with respect to systemic adverse events » (traduction : « les données [des études de pharmacocinétique(21) et pharmacodynamique(22)] ne sont, à elles seules, pas suffisantes pour justifier un avertissement différencié qui donnerait l’impression que Lucentis serait plus sûr que les autres traitements par anti-VEGF concernant les effets indésirables systémiques »).
146. En second lieu, le CHMP a indiqué que le laboratoire Novartis n’avait apporté aucun élément susceptible de modifier son constat, réalisé lors de l’évaluation de la modification du RCP d’Avastin (cf. paragraphes 132 à 139 ci-dessus), selon lequel aucune donnée scientifique ne permettait de considérer que Lucentis et Avastin présentaient des différences dans leur profil de sécurité systémique. Le CHMP a rappelé, sur ce point, que « results of the CATT and IVAN studies have already been assessed by the CHMP in a previous procedure concluding that there was no evidence for a difference in the systemic safety profile of ranibizumab and bevacizumab » (traduction libre : « les résultats des études CATT et IVAN ont déjà été étudiés par le CHMP dans une procédure antérieure, arrivant à la conclusion qu’il n’y avait pas de preuve d’une différence dans le profil de sécurité systémique du ranibizumab [Lucentis] et du bevacizumab [Avastin] »). En conséquence, le CHMP a refusé toute modification du RCP de Lucentis qui permettrait de considérer que celui-ci serait plus sûr que les autres traitements par anti-VEGF : « the CHMP (…) did not accept additionnal wording in the warning that would give rise to the impression that ranibizumab is safer with regards to systemic adverse events as compared to other intravitreal anti-VEGF treatments » (traduction libre : « le CHMP (…) n’a pas accepté la proposition d’ajout additionnel dans l’avertissement qui donnerait l’impression que le ranibizumab [Lucentis] est plus sûr que les autres traitements anti-VEGF intravitréens, s’agissant des effets indésirables systémiques »).
147. En conclusion, compte tenu du fait que l’existence de ce risque théorique était déjà mentionnée dans la section 4.8 « Effets indésirables » du RCP de Lucentis, le CHMP a préconisé de la compléter, en ajoutant les cas de crises cardiaques et d’infarctus du myocarde documentés. Par ailleurs, conformément à ce qui avait été fait lors de la modification du RCP Avastin, le CHMP a recommandé l’insertion, dans la section 4.4 « Mises en gardes spéciales et précautions d’emploi », d’un paragraphe sur l’existence d’effets systémiques indésirables liés à l’utilisation des anti-VEGF en intravitréen.
148. La Commission européenne a modifié sa décision d’AMM en ce sens le 4 juillet 2013.
L’analyse des autorités de santé françaises
Le point d’information de l’AFSSAPS du 10 septembre 2009
149. Le 10 septembre 2009, l’AFSSAPS (devenue, en 2012, l’Agence nationale de sécurité des médicaments ou « ANSM ») a publié un point d’information sur l’utilisation « hors AMM » d’Avastin en ophtalmologie (cotes 48387 et 48388).
150. À cette occasion, l’AFSSAPS a rappelé les données scientifiques disponibles concernant cette utilisation. Elle a par ailleurs indiqué que des essais cliniques étaient en cours, visant à comparer l’efficacité clinique d’Avastin à celle de Lucentis dans le traitement de la DMLA, comme l’étude GEFAL susvisée.
151. Plus spécifiquement, l’AFSSAPS a indiqué que « des incertitudes persistent » sur la stabilité et les conditions d’asepsie liées à l’utilisation d’Avastin pour le traitement de la DMLA, soulignant que « la forme pharmaceutique actuelle d’Avastin n’est pas adaptée à une administration intravitréenne ». Elle a par ailleurs précisé que « les données de sécurité d’emploi d’Avastin en ophtalmologie sont encore limitées », ce qui justifiait notamment la mise en place de l’étude GEFAL ou encore d’études dans d’autres pays européens.
152. Elle en conclut « dans l’attente des résultats de ces études, l’AFSSAPS rappelle l’existence de médicaments autorisés après évaluation pour le traitement de la DMLA et conseille la prudence dans l’utilisation d’Avastin en intravitréen ».
Le point d’information de l’AFSSAPS du 16 septembre 2011
153. Le 16 septembre 2011, l’AFSSAPS a publié un nouveau point d’information sur l’utilisation d’Avastin « hors AMM » en ophtalmologie (cote 49619).
154. Elle y souligne notamment l’existence de « premiers résultats encourageants » des différents travaux de recherches sur l’utilisation d’Avastin dans le traitement de la DMLA. Elle indique ainsi que les résultats à 1 an d’une étude américaine, l’étude CATT susvisée, « montre que l’efficacité du bevacizumab [Avastin] dans la DMLA serait équivalente à celle du ranibizumab (Lucentis) ».
155. Elle précise par ailleurs que le laboratoire Roche, interrogé sur son intention de développer Avastin dans le traitement de la DMLA, « formule toutefois des réserves sur la méthodologie de l’étude américaine et sur la tolérance du produit lors de son utilisation en ophtalmologie ».
La note du directeur général de l’AFSSAPS au directeur général de la santé du 20 mars 2012
156. Le 20 mars 2012, le directeur général de l’AFSSAPS a adressé une note au directeur général de la santé au sujet de l’utilisation « hors AMM » d’Avastin en ophtalmologie.
157. Il indique dans cette note « il ne m’est pas possible d’encourager cette pratique, en raison du risque infectieux et/ou de surdosage lié au reconditionnement et/ou de surdosage lié au reconditionnement des flacons d’Avastin en seringues pour injection intra-oculaire ».
158. Il souligne également, après avoir rappelé les dispositions susvisées de l’article L. 5121-12-1 du code de la santé publique relatives aux RTU, issues de la loi Bertrand (cf. paragraphe 22 ci-dessus), que « prescrire Avastin dans les indications couvertes par l’Autorisation de Mise sur le Marché (AMM) de la spécialité Lucentis ne correspond pas aux termes de la loi ».
159. Il conclut qu’« il n’y a aucun argument scientifique pour préférer l’Avastin par rapport au Lucentis en ophtalmologie ».
La recommandation de la HAS de juin 2012
160. En juin 2012, la HAS a publié une recommandation professionnelle ayant pour objectif d’optimiser la stratégie diagnostique des patients atteint de DMLA et d’homogénéiser les bonnes pratiques de prise en charge de la DMLA (cotes 14358 à 14410).
161. Plus particulièrement, dans son chapitre 4, « Méthodes thérapeutiques disponibles », la HAS recommande un traitement par anti-VEGF, visant plus particulièrement le ranibizumab (Lucentis) et le pegaptanib (Macugen), qui disposaient tous deux d’une AMM pour le traitement de la DMLA, et l’aflibercept (Eylea) dont la demande d’AMM était en cours d’évaluation en Europe pour cette indication.
162. La HAS mentionne également l’utilisation « hors AMM » du bevacizumab (Avastin) en ophtalmologie. Sur ce point, la HAS indique que les résultats à un et à deux ans de l’étude CATT susvisée, confirmés par les résultats à 1 an de l’étude IVAN susvisée, démontrent « une efficacité du bevacizumab [Avastin] non inférieure à celle du traitement de référence [Lucentis] » et qu’« il n’y a pas de différence statistiquement significative en termes de tolérance à deux ans entre les deux molécules sur le risque de décès, d’accidents thromboemboliques ». Elle souligne en revanche que « la proportion observée de patients avec un ou plusieurs effets indésirables graves systémiques est plus importante avec le bevacizumab [Avastin] que pour le ranibizumab [Lucentis] », précisant néanmoins que « ce résultat est à interpréter avec prudence en raison de la diversité des pathologies rapportées dans l’étude CATT ».
163. La HAS relève donc qu’« à ce jour, il n’existe pas de données publiées à partir d’études randomisées(23) comparatives de haut niveau de preuve mettant en évidence une mauvaise tolérance du bevacizumab [Avastin] à long terme dans le traitement des néovaisseaux choroïdiens(24) de la dégénérescence maculaire liée à l’âge », tout en invitant à « rester vigilant ».
164. En conclusion, la HAS recommande de se référer au point d’information de l’AFSSAPS (devenue ANSM) de septembre 2009 susvisé, soulignant que celle-ci « reste sur une position de prudence et rappelle les incertitudes de cet usage au regard de la sécurité des patients ».
L’instruction de la DGS de juillet 2012
165. Le 11 juillet 2012, le directeur général de la santé a adressé aux directeurs régionaux des agences régionales de santé une instruction relative à la législation applicable aux préparations magistrales et notamment aux préparations de seringues pour injection intravitréenne de la spécialité pharmaceutique Avastin (cotes 1574 à 1576).
166. Cette instruction indique que « des cas d’endophtalmie (infection de l’œil) ont été signalés dans la littérature scientifique et médicament ainsi que via le système de pharmacovigilance » et que « des publications récentes ont confirmé les risques liés à l’utilisation d’Avastin en ophtalmologie ».
167. Elle en conclut que « la préparation de seringues par répartition aseptique d’une solution de bevacizumab (Avastin), pour injection intravitréenne, est interdite, compte tenu de l’existence d’une spécialité adaptée (Lucentis, ranibizumab) possédant une AMM pour le traitement de la DMLA et spécifiquement formulée et présentée pour les injections intravitréennes ».
168. Cette instruction a été abrogée après la publication de la RTU Avastin (cf. paragraphe 72 ci-dessus), avec la publication d’une note d’information de la DGS du 31 août 2015(25).
L’instruction de la DGS d’août 2012
169. Le 10 août 2012, le directeur général de la santé a adressé aux directeurs régionaux des agences régionales de santé une instruction ayant pour objet de compléter et modifier l’instruction de juillet 2012 (cote 48524 et 48525).
170. Cette instruction revient sur les conditions dans lesquelles Avastin peut être utilisé, « pour ce qui concerne les autres indications oculaires pour lesquelles Lucentis ni aucun autre médicament n’a d’AMM ».
171. Soulignant que l’ANSM a été saisie en vue de l’élaboration d’une RTU pour Avastin, l’instruction conclut donc que « dans l’attente de cette RTU, la prescription d’Avastin est possible dans le respect des conditions du 2° du I et du III de l’article L. 5121-12-1 [du code de la santé publique] précité ».
4. L’EVOLUTION DES VENTES DES PRODUITS CONCERNES EN VILLE ET A L’HOPITAL
172. Il convient de revenir sur l’évolution des ventes d’Avastin (a) et de Lucentis (b) en ophtalmologie.
a) Les événements ayant eu un impact sur l’utilisation par les médecins ophtalmologistes d’Avastin en ophtalmologie
173. Comme décrit ci-dessus, l’usage « hors AMM » d’Avastin en ophtalmologie s’est développé, à l’origine, de manière ad hoc, en dehors de tout cadre réglementaire et dans un contexte marqué par la rareté des publications scientifiques sur le sujet.
174. Plusieurs événements ont eu un impact sur l’utilisation d’Avastin en ophtalmologie.
175. En premier lieu, en 2007, l’AMM accordée à Lucentis et l’admission de cette spécialité au remboursement à 100 % par la sécurité sociale (cf. paragraphes 77 à 79 ci-dessus) ont diminué les incitations des médecins ophtalmologistes à recourir à Avastin.
176. En effet, dès lors qu’Avastin était utilisé « hors AMM », il n’était pas remboursé, et son coût devait être intégré dans la tarification de la consultation et/ou de l’hospitalisation de l’établissement de santé. À l’inverse, en prescrivant Lucentis, que les patients pouvaient acquérir en officine de ville, moyennant un remboursement total par la sécurité sociale, et apporter le jour de l’injection, les médecins limitaient les coûts d’approvisionnement de leur établissement. Plusieurs médecins hospitaliers ont ainsi déclaré avoir recours à cette méthode, selon laquelle le patient qu’ils suivaient allait acheter Lucentis « en ville », pour qu’ils le lui administrent ensuite à l’hôpital. À titre d’illustration, un praticien hospitalier au CHU de Lille a ainsi indiqué : « Lors du diagnostic, on précise le schéma thérapeutique qui comprend souvent 3 injections consécutives à un mois d’intervalle. La prescription se fait sur une ordonnance de médicament d’exception pour du Lucentis ou de Eylea. Le patient se rend ensuite dans sa pharmacie de ville pour se procurer le médicament. Ce dernier est pris en charge à 100 % par la sécurité sociale » (cote 13595). De même, le chef de service en ophtalmologie au CHNO des Quinze-Vingt et à la fondation ophtalmologique Rothschild a déclaré : « Une fois diagnostiqué, si nous estimons qu’une injection est nécessaire, nous envoyons le patient se fournir en Lucentis ou en Eylea dans la pharmacie de son choix et nous fixons son rendez-vous pour l’injection. Dans ce cas, nous ne facturons que l’injection et pas le produit » (cote 13585).
177. Ainsi, certains médecins ophtalmologistes ont déclaré ne plus utiliser Avastin depuis l’arrivée de Lucentis sur le marché et son admission au remboursement. En revanche, Avastin a continué d’être largement utilisé pour les autres indications (OMD, OVR, etc.). À titre d’illustration, le chef de service en ophtalmologie au CHNO des Quinze-Vingt et à la fondation ophtalmologique Rothschild a déclaré : « Quand Lucentis est arrivé et a obtenu son remboursement nous avons considéré que nous ne pouvions pas continuer à utiliser Avastin pour les indications pour lesquelles Lucentis avait l’AMM. En revanche, nous avons continué à utiliser Avastin dans les autres indications qui se sont rétrécies progressivement » (cote 13584 ; voir également cote 13594).
178. En second lieu, l’instruction de la DGS de juillet 2012 a conduit la plupart des médecins ophtalmologistes à cesser leur utilisation d’Avastin (cf. paragraphes 165 à 168 ci-dessus). Toutefois, comme l’a souligné l’ANSM dans le cadre du contentieux devant le Conseil d’État sur la RTU Avastin, l’usage d’Avastin « hors AMM » dans le traitement de la DMLA a continué, nonobstant l’adoption de cette instruction (cote 15040 : « compte tenu de la persistance de l’utilisation de la spécialité Avastin dans le traitement de la DMLA en dépit de l’instruction précitée du 11 juillet 2012 »).
179. Cette instruction a en effet provoqué la surprise de nombreux médecins ophtalmologistes (cotes 13586 et 13718). Le syndicat national des pharmaciens des établissements publics de santé a d’ailleurs adressé à M. L…, directeur général de la santé, un courrier le 6 août 2012, afin de faire part de son incompréhension (cotes 14356 et 14357). Outre la contestation de la justification scientifique de cette instruction, son champ particulièrement large a été critiqué. En effet, les réserves d’utilisation conduisaient à interdire de façon générale tout usage d’Avastin, en ce compris pour les autres indications oculaires, pour lesquelles ni Lucentis, ni aucun autre médicament, n’avait d’AMM. Ces critiques ont donné lieu, un mois à peine après l’adoption de l’instruction du 11 juillet 2012, à l’instruction rectificative du 10 août 2012 (cf. paragraphes 169 à 171 ci-dessus).
180. En pratique, l’utilisation d’Avastin dans le traitement de la DMLA a connu une diminution constante entre 2008 et 2013. Ainsi, selon les estimations internes de Novartis, sa part de marché a décru très rapidement entre 2008 et 2010, passant de 15 % en janvier 2008 à 6 % en novembre 2009 (source : Panel Rekam ; cote 13977). En mai 2010, la part de marché d’Avastin était toujours de 6 %, décomposée de la façon suivante : entre 13 % et 17 % dans les établissements publics et environ 2,7 % dans les établissements privés (source : Panel Rekam, cote 13978). Par la suite, la part de marché d’Avastin est passé de 6 % à 3 % entre octobre 2010 et décembre 2012, la diminution la plus marquée ayant eu lieu en juillet 2012 (source : Baromètre Avastin ; cote 4705).
Évolution de la part de marché d’Avastin dans le traitement de la DMLA, selon les estimations internes de Novartis précitées
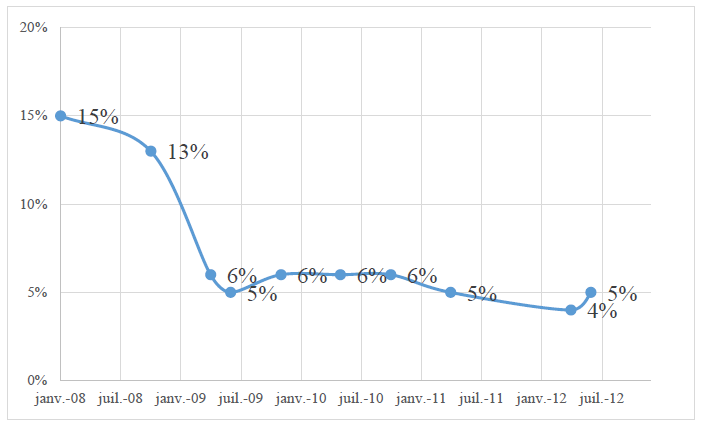
Source : Novartis
b) Les événements ayant eu un impact sur l’utilisation par les médecins ophtalmologistes de Lucentis en ophtalmologie
181. Dès son arrivée sur le marché, Lucentis a atteint des parts de marché très importantes pour le traitement de la DMLA. Ainsi, selon les estimations internes de Novartis, en juillet 2009, la part de marché de Lucentis s’élevait à 86 %, loin devant les spécialités concurrentes (Avastin (6 %), Vysudine (5 %) et Macugen (2 %)) (source : Panel Rekam ; cote 2368).
Part de marché de Lucentis et de ses concurrents dans le traitement de la DMLA en juillet 2009, selon les estimations internes de Novartis
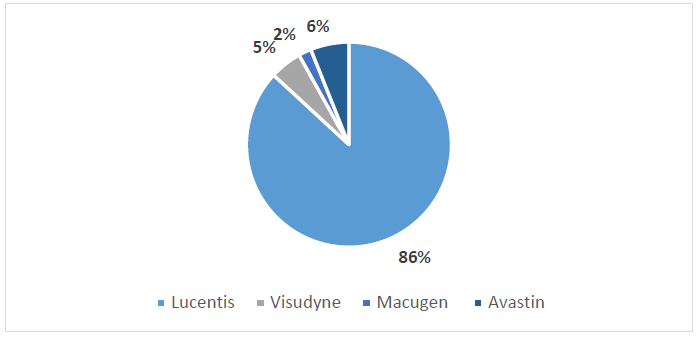
Source : Novartis
182. Entre octobre 2010 et décembre 2012, elle a oscillé autour de 90 %, allant jusqu’à 93 % en décembre 2012 (source : Baromètre Avastin ; cote 4705).
183. Après son entrée sur le marché en novembre 2013, Eylea a capté 36 % des parts de marché en trois mois (source : données GERS, cote 45129). La pénétration d’Eylea sur ce marché a donc été particulièrement rapide.
C. LES ACTEURS CONCERNES
184. Seront présentés successivement les laboratoires pharmaceutiques concernés et leurs relations (1), puis les relations entre laboratoires et professionnels de santé (2).
1. LES RELATIONS ENTRE LES LABORATOIRES CONCERNES
185. Il convient de présenter les laboratoires concernés (a) et leurs relations contractuelles et capitalistiques (b).
a) Les laboratoires concernés
Le laboratoire Genentech
186. Genentech Inc. est une société de droit américain, fondée en 1976, spécialisée dans le développement de médicaments issus des biotechnologies, ayant son siège social à San Francisco.
187. Comme indiqué aux paragraphes 63 et 76 de la présente décision, Genentech, qui a développé Avastin et Lucentis, commercialise ces deux médicaments sur le territoire américain. Le laboratoire a en revanche confié la commercialisation dans le reste du monde de ces produits, d’une part, au laboratoire Roche concernant Avastin et, d’autre part, au laboratoire Novartis, concernant Lucentis.
188. Depuis 1990, Genentech est détenue à 60 % par la société Roche Holding AG, dont la participation est contrôlante. Depuis le 26 mars 2009, la société Roche Holding AG détient l’intégralité du capital social de Genentech (cote 16749).
Le laboratoire Novartis
189. La société Novartis AG, cotée à la bourse suisse (« SIX Swiss Exchange ») et à la bourse de New-York, dont le siège social est situé à Bâle en Suisse, est la société mère faîtière du groupe Novartis (cote 15490).
190. Les filiales du groupe Novartis établies en France sont les sociétés Novartis Groupe France SA (RCS 709 804 538), Novartis Pharma SAS (RCS 410 349 070), Sandoz SAS (RCS 552 123 341) et Laboratoire Alcon SAS (RCS 652 009 044). La société Novartis Groupe France, qui est détenue à 100 % par la société Novartis AG, détient 100 % de la société Novartis Pharma.
191. La société Novartis Pharma est organisée en deux unités d’affaires, « Oncologie » et « Produits pharmaceutiques », qui constituent ensemble la division « Médicaments innovants ». Chaque unité d’affaires est divisée en structures dénommées « franchises », une pour chaque domaine thérapeutique. La franchise « Ophtalmologie », qui appartient à l’unité « Produits pharmaceutiques », est en charge de la commercialisation de Lucentis pour l’ensemble de ses indications en France.
Le laboratoire Roche
192. Fondé en 1896 par Fritz Hoffmann et Adèle La Roche, le groupe Roche est une entreprise pharmaceutique internationale.
193. La société Hoffmann-La Roche AG, basée en Suisse, est la principale société opérationnelle du groupe Roche. Elle est détenue intégralement par la société Roche Holding AG, société faîtière du groupe Roche, cotée dans l’indice SIX Swiss Exchange de la bourse de Zurich, dont le siège social est situé à Bâle, en Suisse.
194. Au 31 décembre 2017, la société Roche Holding AG était contrôlée par un groupe d’actionnaires composé notamment de membres des familles Hoffmann et Oeri, détenant ensemble 45,01 % des actions conférant un droit de vote. Ce groupe d’actionnaires exerce en commun les droits de vote conférés par ces actions, conformément à un accord en vigueur depuis 1948 (cotes 16747 à 16748).
195. L’autre principal actionnaire de Roche est le groupe Novartis, qui détient un peu moins d’un tiers des actions. Plus précisément, Novartis détient 6,2 % du capital (les actions sans droit de vote représentant 81,5 % du capital de Roche Holding) et 33,33 % des droits de vote de Roche Holding (cote 47658).
196. Le groupe Roche est présent en France via ses activités (i) de pharmacie, à travers la société Roche SAS (RCS 552 012 031), (ii) de diagnostic, à travers la société Roche Diagnostics France SASU (RCS 380 484 766) et (iii) de gestion du diabète, à travers la société Roche Diabètes Care France SASU (RCS 800 418 493).
197. La société Roche SAS, filiale de distribution, est en charge de la commercialisation d’Avastin en France. Elle est détenue indirectement à 100 % par la société Roche Holding AG, par l’intermédiaire de la société Roche Finanz AG (cote 16749).
b) Les relations contractuelles entre Genentech, Roche et Novartis pour la commercialisation d’Avastin et Lucentis
198. Comme indiqué aux paragraphes 63 et 76 de la présente décision, Genentech a confié la commercialisation d’Avastin au laboratoire Roche et de Lucentis au laboratoire Novartis dans le monde entier (à l’exception des États-Unis).
Le contrat de licence entre Genentech et Roche pour Avastin
199. Les relations entre Genentech et Roche pour la commercialisation d’Avastin sont régies par un accord cadre, entré en vigueur en 1995, remplacé en 1999 par un nouvel accord, qui accorde à Roche le bénéfice d’une option, produit par produit, pour la licence et la commercialisation exclusive des produits de Genentech, en cours de développement ou prochainement en développement, dans le monde entier, sauf aux États-Unis (cote 16750).
200. Roche a exercé son droit d’option concernant Avastin le 23 juin 2003. Depuis lors, Roche détient l’exclusivité de la commercialisation d’Avastin en dehors des États-Unis. Sur cette zone géographique, Roche est seule responsable des relations avec les autorités de santé, en vue des enregistrements et autorisations des produits pour lesquels elle a exercé son droit d’option. Elle est ainsi titulaire des AMM délivrées pour Avastin dans cette zone géographique (cotes 16750 et 45042).
201. Par ailleurs, l’accord cadre prévoit la mise en place de quatre comités, dans lesquels Roche et Genentech sont représentés à parité (cotes 45041 et 45042).
202. Tout d’abord, au sein du « Joint Commercialization Committee » (ou « Comité de commercialisation conjoint »), Genentech communique à Roche des informations sur les produits en cours de développement, et notamment le résultat des études cliniques nécessaires au dépôt d’une demande d’AMM, afin notamment de permettre à Roche de décider s’il souhaite ou non exercer son option sur lesdits produits. Roche communique, quant à lui, via ce comité, des informations à Genentech concernant les produits qu’elle a en licence.
203. Ensuite, le « Management Committee » (ou « Comité de gestion ») se réunit, au moins une fois par an, pour discuter du statut de développement et de commercialisation de l’ensemble des produits objet de l’accord cadre, ainsi que de tout autre sujet porté à son attention par un des trois autres comités.
204. Puis, le « Development Committee » (ou « Comité de développement ») se réunit, au moins trois fois par an, pour discuter du développement des produits pour lesquels Roche a exercé son droit d’option. Ce comité est en charge de superviser et coordonner les efforts de développement au niveau mondial desdits produits. L’article III.3 du contrat prévoit en particulier que la conception de la stratégie mondiale de développement relève principalement de Genentech, tandis que les décisions relatives aux efforts de développement et à l’enregistrement dans le territoire concédé relèvent uniquement de Roche (cote 50402).
205. En pratique, il est prévu que ce comité se réunisse pour proposer un « plan de développement » pour chaque produit au niveau mondial. À ce sujet, les représentants de Roche ont indiqué en audition : « Un plan de développement se construit très en amont, en fonction de ce que l'on imagine du potentiel de la molécule dans des indications. Pour ce qui est d'Avastin, le plan de développement remonte forcément plusieurs années avant la première AMM, et donc en l'espèce dans les années 90. Ce plan concernait uniquement l'oncologie, puisque la force de Genentech était sur l'oncologie. C'était l'une des premières molécules avec ce mode d'action en oncologie […] Pour tous les pays hors Etats-Unis on a un Lifecycle Leader [responsable du cycle de vie] qui organise, qui orchestre, qui s'occupe de la cohérence entre les plans de développement, la commercialisation, en somme toute la vie du produit. Nous avons autour de lui des équipes dédiées, notamment médicales et d'accès (« Market Access »). Ce Lifecycle Leader [responsable du cycle de vie] est quelqu'un de très transversal » (cote 16737).
206. Par ailleurs, Roche doit communiquer à Genentech, avant le 30 novembre de chaque année, les informations suivantes : (i) le statut et les projets d’essais cliniques nécessaires à l’enregistrement du produit, (ii) le statut et les projets d’enregistrement du produit en vue de sa commercialisation, (iii) les ventes prévisionnelles du produit concerné, et (iv) un résumé des opérations promotionnelles et des études de marché (cote 50400). Genentech est donc régulièrement informé de la politique commerciale et du positionnement d’Avastin dans le territoire concédé à Roche.
207. Enfin, le « Joint Finance Committee » (ou « Comité financier conjoint ») se réunit en tant que de besoin pour discuter des questions financières en lien avec l’accord cadre.
208. L’accord organisant les relations entre Genentech et Roche, en particulier pour l’exploitation d’Avastin, confère à Roche une certaine autonomie, notamment s’agissant des relations avec les autorités d’enregistrement en dehors des États-Unis, et des actions de promotion et de commercialisation.
209. Toutefois, cette autonomie intervient dans un cadre très contraint. Tout d’abord, les relations entre Genentech et Roche sont organisées suivant un schéma dans lequel Genentech est responsable de l’essentiel du développement médical et scientifique des produits. En outre, les actions de Roche doivent demeurer dans le cadre du plan de développement négocié avec Genentech, qui inclut en particulier les indications envisagées pour chacun des produits. Enfin, les obligations de remontées d’informations qui pèsent sur Roche, ainsi que le fonctionnement des quatre comités décrits ci-dessus, permettent de très nombreux contacts entre les deux entreprises.
Le contrat de licence entre Genentech et Novartis pour Lucentis
210. Les relations entre Genentech et Novartis pour l’exploitation de Lucentis sont régies par un accord de licence et de collaboration conclu le 20 juin 2003 pour le développement et la commercialisation de produits pharmaceutiques contenant le ranizumab, vendu par Novartis sous la marque Lucentis dans le monde entier, à l’exception des États-Unis (cote 45137).
211. Cette licence est accordée uniquement pour un domaine spécifique, en l’occurrence le traitement des maladies de l’œil et/ou de la rétine et/ou de la vascularisation choroïde (cote 45138).
212. Novartis et Genentech sont seules responsables des activités réglementaires et de la commercialisation de Lucentis sur leur territoire respectif (cote 15532). Toutefois, chacune des parties est tenue d’informer l’autre des demandes des autorités de santé compétentes sur son territoire (cote 45140).
213. Par ailleurs, l’accord entre Genentech et Novartis a institué un « Joint Management Committee » (ou « Comité de gestion conjoint ») et une « Joint Project Team » (ou « Equipe projet conjoint »), au sein desquels les deux parties sont représentées de manière égale.
214. Plus particulièrement, le « Joint Management Committee » établit des plans de développement et des plans pour le cycle de vie du produit et gère sur cette base le développement conjoint de Lucentis dans le monde entier. Il détermine par ailleurs la partie responsable de la conception et de la conduite des essais cliniques relatifs à la DMLA et d’autres indications pour Lucentis. En outre, il supervise le positionnement commercial de la marque Lucentis dans le monde (cote 45139).
215. Plus précisément, le plan de développement défini de façon conjointe est un plan complet pour le développement mondial conjoint des produits sous licence dans le « Champ », correspondant au traitement des maladies de l’œil et/ou de la rétine et/ou de la vascularisation choroïde (cotes 15530 et 45138). L’accord précise que Novartis ne peut pas organiser ou soutenir des études pour des indications en dehors du « Champ » (cote 15530).
216. Enfin, l’accord prévoit un grand nombre d’obligations d’information et de consultation entre Novartis et Genentech, sur tous les différents aspects de la vie du produit (cotes 45114 et 45115).
217. L’article 5.1 e) de l’accord prévoit, en particulier, que chaque partie devra informer l’autre des communications intervenues avec les autorités de santé, et lui donner la possibilité de participer aux rencontres d’importance significative (cotes 15532 et 15533).
218. De même, l’article 6.4 de l’accord prévoit que le « Joint Management Committee » doit contrôler les principaux messages de promotion diffusés tant par Novartis que par Genentech, et tous les changements qui pourraient être proposés pour ces messages. En outre, les deux entreprises doivent travailler ensemble, de bonne foi, pour maintenir l’alignement et la cohérence du positionnement commercial du produit dans le monde entier (cote 15535).
219. Enfin, le « Joint Management Committee » doit également revoir tout article scientifique, présentation ou intervention dans un colloque de Novartis ou de Genentech en lien avec le produit sous licence. Cette obligation doit également s’étendre, dans la mesure du possible, à tout article ou intervention d’un partenaire ou d’un chercheur financé par Novartis (cote 15560).
220. En conclusion, l’accord conclu entre Genentech et Novartis organise une coopération très étroite entre les deux entreprises, qui définissent ensemble, pour Lucentis, un plan de développement, et coordonnent les messages commerciaux, les communications scientifiques, ainsi que les relations avec les autorités de santé. Enfin, les obligations de remontées d’informations et le fonctionnement du « Joint Management Committee » décrit ci-dessus permettent de très nombreux contacts entre les deux entreprises.
c) Schéma récapitulatif des liens capitalistiques et contractuels entre Genentech, Roche et Novartis
221. Les relations capitalistiques et contractuelles existant entre Genentech, Roche et Novartis peuvent être résumées dans le schéma suivant :
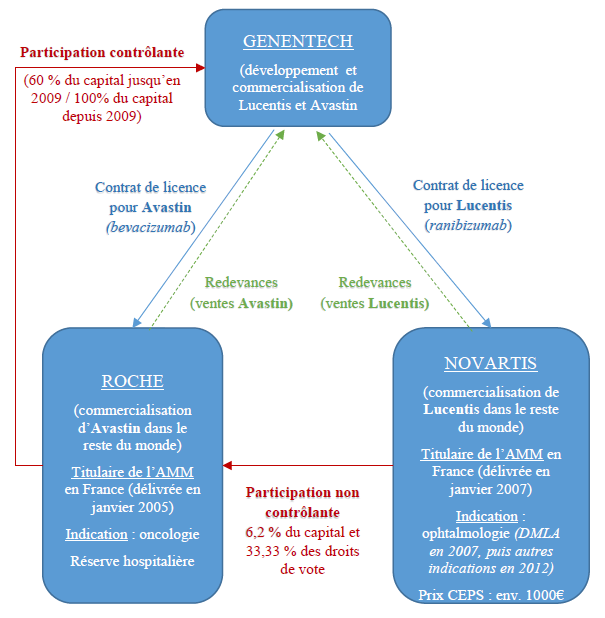
2. LES RELATIONS ENTRE LES LABORATOIRES PHARMACEUTIQUES ET LES PROFESSIONNELS DE SANTE
222. Dans le secteur du médicament, et notamment lorsqu’il s’agit de médicaments remboursables, la décision d’utiliser une spécialité ou une autre n’est pas prise par le patient concerné, mais par un professionnel de santé. Par conséquent, indépendamment du fait que la publicité auprès du grand public pour les médicaments soumis à prescription obligatoire est interdite en France, un laboratoire pharmaceutique, qui souhaite développer ses ventes est souvent conduit à entretenir des contacts avec les professionnels de santé, ces derniers constituant les prescripteurs de l’usage des médicaments.
223. Toutefois, en vue de limiter les stratégies de communication des laboratoires pharmaceutiques et d’assurer le respect de la déontologie des médecins, les interactions entre les laboratoires pharmaceutiques et les professionnels de santé sont strictement réglementées (a). Les laboratoires recourent ainsi souvent à des méthodes indirectes d’influence, notamment au travers d’actions visant les « Key Opinion Leaders » (ou leader d’opinion de premier plan, ci-après, « KOL ») (b).
a) Les règles relatives à la promotion du médicament auprès des professionnels de santé
224. Les règles applicables pour la promotion du médicament en France ont été compilées et synthétisées par la HAS, dans un document de janvier 2013, intitulé « Description de la régulation de la promotion des produits de santé en France »(26).
225. Ce document indique tout d’abord que « tous les produits de santé sont soumis aux dispositions du code de la consommation qui interdit les pratiques commerciales trompeuses (articles L. 121-1 et suivants). Une pratique commerciale (publicité par exemple) est notamment trompeuse lorsqu’elle repose sur des allégations, indications ou présentations fausses ou de nature à induire en erreur ».
226. Concernant la promotion des médicaments auprès des professionnels de santé, « depuis la loi du 29 décembre 2011 (loi n° 2011-2012) la publicité pour les médicaments auprès des professionnels de santé est soumise à un contrôle a priori au terme duquel une autorisation, dénommée visa PM, est délivrée par l’ANSM. Les firmes pharmaceutiques déposent donc les supports promotionnels qu’elles souhaitent utiliser auprès de l’ANSM qui délivre ou non une autorisation (article L. 5122-9 du CSP) ».
227. Le code de la santé publique contient également un ensemble de dispositions limitant fortement la possibilité pour les entreprises pharmaceutiques d’offrir des cadeaux ou de rémunérer des professionnels de santé (voir notamment, les articles L. 4113-6 et L. 5122-10 du code de la santé publique).
228. Par ailleurs, le LEEM et le CEPS ont conclu, en 2004, une charte de la visite médicale dont l’objectif principal est de « renforcer le rôle de la visite médicale dans le bon usage des médicaments remboursés aux assurés sociaux et la qualité de l’information délivrée ». Cette charte contient un certain nombre de prescriptions relatives à la qualité de l’information transmise par le délégué médical, concernant notamment la publicité comparant deux ou plusieurs médicaments ou encore les études scientifiques postérieures à la délivrance de l’AMM.
229. En octobre 2014, le LEEM et le CEPS ont signé une nouvelle « Charte de l’information promotionnelle ». Ce document complète les informations susvisées, en indiquant notamment que « Lorsque l’entreprise utilise de telles études, elle les présente de façon complète et impartiale ».
b) Les relations des laboratoires pharmaceutiques avec les « Key Opinion Leaders »
230. Selon les termes d’une présentation effectuée par un cabinet de consultant, un « KOL » est un professionnel de santé bénéficiant d’une reconnaissance significative et d’une compétence d’expert reconnu dans son domaine. Cette notoriété est généralement due à la fois au statut professionnel du praticien (notamment lorsque celui-ci occupe un poste hospitalo-universitaire dans un service hospitalier important) et au nombre d’articles et d’études publiés par ses soins (cote 14311).
231. En 2013, la HAS a publié sur son site Internet un manuel pratique intitulé « Comprendre la promotion pharmaceutique et y répondre »(27). Ce manuel fournit des informations sur les raisons poussant l’industrie pharmaceutique à s’attacher le concours des leaders d’opinion de premier plan (ou « KOL »).
232. Ainsi, « Les médecins ne sont pas réceptifs aux campagnes classiques de vente et de marketing. Mais ils sont réceptifs aux opinions de leurs confrères. En fait, certains médecins ont un immense pouvoir de persuasion sur leurs pairs. Ces médecins ont gagné le respect et l'attention des autres prescripteurs et ont été reconnus pour leur expertise et connaissance des thérapies innovantes et émergentes. Mais surtout, ils sont à même d'essayer, d'adopter et de préconiser ces nouveaux produits ... D'un point de vue marketing, ces médecins sont au sommet de la hiérarchie médicale – ils sont les leaders d'opinion de premier plan par excellence ». De même, « une présentation faite à des confrères par un médecin éminent a moins l'air d'être une promotion qu'une présentation faite par un employé d'une entreprise pharmaceutique. Le succès de cette technique d'influence est peut-être dû en partie au fait que l'auditoire n'a pas conscience que cela fait partie d'une campagne promotionnelle pour un produit ».
233. Afin de développer les relations avec les KOL, les laboratoires pharmaceutiques ont ainsi établi en interne une fonction spécifique, celle de « Medical Scientific Liaison » (ou Référent Médical, ci-après « MSL »). Cette personne, qui est généralement elle-même médecin, biologiste ou un autre professionnel de santé, a pour rôle d’animer les relations avec les médecins et de proposer des partenariats à ces derniers.
234. Plus spécifiquement, l’industrie pharmaceutique a mis en place un ensemble de techniques permettant d’élaborer une cartographie des KOL d’un secteur donné, en fonction de leur degré de notoriété, de leur compatibilité avec le discours prôné par l’entreprise et de leur inclinaison à participer à des événements financés par l’industrie. Ainsi, des cabinets de consultants ont pu proposer, par exemple, une structuration des « niveaux » de KOL en pyramide (les KOL les plus importants – qui se trouvent tout en haut de la pyramide – sont ceux ayant une influence sur les autorités de santé et les pouvoirs publics en général, cotes 14312 et 14330) ou encore sous la forme d’une matrice (identifiant les partisans et les opposants au médicament concerné et leur niveau d’influence sur les autres prescripteurs, cote 14321).
235. Un document interne de Novartis, intitulé « M3PH Medical plan 2011 / SciOps », fournit un descriptif des ressources utilisées par Novartis dans ses relations avec les KOL (cotes 14892 à 14904). Dans le domaine de la DMLA, 93 KOL ont été identifiés par le laboratoire, comme devant être régulièrement visités par les deux référents médicaux ou « MSL » (cote 14895).
La présentation contient également un plan de gestion des KOL en pyramide comme présenté ci-dessus (cote 14896). Enfin, le document fait état d’une méthode d’évaluation des positions et de l’influence des KOL, par le biais d’une moyenne pondérée attribuée à ces derniers (cotes 14900 et 14901) :
D. L’AFFAIRE ITALIENNE
236. Le 27 février 2014, l’Autorita Garante della Concorrenza e del Mercato (ci-après, « l’AGCM ») a adopté une décision infligeant aux laboratoires Roche et Novartis une sanction pécuniaire totale de 182,5 millions d’euros, pour avoir mis en place une entente anticoncurrentielle « par objet », contraire à l’article 101, paragraphe 1, du TFUE, en vue de générer une différence artificielle entre les spécialités Avastin et Lucentis pour le traitement de la DMLA.
237. Les pratiques mises en œuvre ont consisté en la diffusion d’un discours de nature à générer une crainte parmi les professionnels de santé, les autorités de santé et le grand public, en exagérant les risques découlant de l’usage intravitréen d’Avastin. Selon l’AGCM, Roche et Novartis ont cherché, en dissuadant l’usage « hors AMM » d’Avastin, à accroître les ventes du produit le plus cher (Lucentis). L’AGCM a considéré que, par les pratiques en cause, les deux groupes ont pu maximiser leur profit respectif, ces comportements étant constitutifs d’une entente anticoncurrentielle par objet.
238. Roche et Novartis ont contesté cette décision devant le Tribunale administrativo regionale per il Lazio, qui a rejeté leur recours (T.a.r. Lazio, 2 décembre 2014, n° 12168). Les deux entreprises se sont pourvues devant le Consiglio di Stato, qui a décidé de sursoir à statuer et de poser des questions préjudicielles à la Cour de justice de l’Union européenne (Consiglio di Stato, ordonnance n° 966 du 11 mars 2016). La Cour de justice a été amenée à se prononcer, notamment, sur la question de savoir si un médicament disposant d’une AMM dans une indication thérapeutique et un médicament utilisé « hors AMM » pour cette même indication pouvaient être considérés comme substituables et donc, relever du même marché pertinent, ou encore sur la question de savoir si une entente portant, dans un contexte d’incertitude scientifique, sur la diffusion d’informations trompeuses sur les effets indésirables de l’utilisation d’un médicament pour le traitement d’une pathologie non couverte par son AMM, pouvait être qualifiée de restriction de la concurrence « par objet » au sens de l’article 101, paragraphe 1, du TFUE.
239. La Cour de justice a répondu aux questions préjudicielles par un arrêt rendu le 23 janvier 2018 (aff. C-179/16, F. Hoffmann-La Roche). En particulier, la Cour de justice a jugé que l’entente entre les groupes pharmaceutiques Roche et Novartis visant à réduire les utilisations ophtalmologiques du médicament Avastin et à accroître celles du Lucentis pouvait constituer une restriction de la concurrence « par objet », après avoir précisé que l’AGCM pouvait considérer que Lucentis et Avastin relevaient du même marché et pouvaient être considérés comme des produits concurrents. Puis le Consiglio di Stato, statuant à nouveau sur le pourvoi de la société Hoffmann-La Roche, a confirmé le jugement du tribunal administratif régional du Lazio et la décision de l’AGCM, par un arrêt du 15 juillet 2019 (Consiglio di Stato, 15 juillet 2019, n° 04990/2019).
240. Plus spécifiquement, le Consiglio di Stato a jugé que la définition du marché pertinent, effectuée par l’AGCM, était correcte, dans la mesure où la réglementation européenne n’interdit pas l’utilisation de médicaments « hors AMM » ni leur reconditionnement pour une telle utilisation. En conséquence, dans la mesure où Avastin était fréquemment prescrit « hors AMM » dans le domaine pharmaceutique, Avastin et Lucentis pouvaient être considérés comme substituables et donc, appartenant au même marché pertinent. Le Consiglio di Stato a par ailleurs confirmé que l’entente entre Roche et Novartis, qui ne saurait être considérée comme une restriction accessoire à l’accord de licence portant sur les médicaments en cause, constituait une restriction de la concurrence par objet. Précisant qu’il n’était pas nécessaire que la preuve scientifique de l’équivalence thérapeutique entre Avastin et Lucentis dans le domaine ophtalmique soit apportée, le Consiglio di Stato a jugé que, même dans un contexte d’incertitude scientifique, il n’était pas permis à deux entreprises concurrentes de diffuser des informations trompeuses, visant à fausser la perception des risques d’un médicament (Avastin) afin d’en réduire l’usage.
E. LES PRATIQUES CONSTATEES
241. Seront successivement exposées les actions entreprises par Novartis auprès des médecins ophtalmologistes, des associations de patients, du grand public et des autorités de santé (1) puis celles mises en œuvre par Novartis, Roche et Genentech auprès des pouvoirs publics (2).
1. LES PRATIQUES MISES EN ŒUVRE PAR NOVARTIS AUPRES DES MEDECINS OPHTALMOLOGISTES, DES ASSOCIATIONS DE PATIENTS, DU GRAND PUBLIC ET DES AUTORITES DE SANTE
242. Le laboratoire Novartis a développé auprès des professionnels de santé – et, plus particulièrement, des médecins ophtalmologistes – mais également auprès des associations de patients, du grand public et des autorités de santé, un discours insistant sur les risques liés à l’utilisation « hors AMM » d’Avastin en ophtalmologie, et plus particulièrement pour le traitement de la DMLA, en contraste avec la sécurité et la tolérance de Lucentis pour un même usage. Ce discours s’est adapté à l’évolution du contexte scientifique et réglementaire exposé ci-dessus (cf. paragraphes 92 à 171 ci-dessus).
a) Les éléments de communication au cours de l’année 2008
Le courriel du 10 mars 2008
243. Dans un courriel du 10 mars 2008, le directeur du marketing en ophtalmologie du groupe Novartis a transmis plusieurs documents aux salariés du groupe en charge du produit Lucentis (cotes 3629 à 3631).
244. Un des documents annexés, intitulé « Avastin Overview Deck » présente les points de différenciation entre Avastin et Lucentis et une analyse des études scientifiques concernant l’efficacité et la sécurité d’Avastin en ophtalmologie (cotes 3635 à 3650).
245. Ce courriel précise que ces informations pourront être utilisés par les salariés de Novartis et les KOL, en faveur de Lucentis : « the document also reviews and critiques some of the supporting data for Avastin and provides an update on the H2H trials. The content in this deck can be used by Novartis medical/scientific staff as well as KOLS and other advocates to support the difference between the two products and defend against the common argument that both products are ‘essentially the same’ » (traduction libre : « le document analyse et critique certaines des données en faveur d’Avastin et fournit une mise à jour des études H2H [de comparaison]. Le contenu de ce document peut être utilisé par le personnel médical et scientifique de Novartis, ainsi que par les KOL et toute autre personne, afin de défendre les différences entre les deux produits et contre l’argument selon lequel les produits seraient ‘essentiellement les mêmes’ », cote 3630).
246. Ces documents ont servi à Novartis pour préparer un document de questions-réponses (« Q&A Lucentis-Avastin », cote 3629).
La présentation du 17 mars 2008 « Lucentis : Major Issues & Strategies »
247. Dans un courriel du 19 mars 2008, le directeur des relations aux publics de Novartis a transmis une présentation du 17 mars 2008 réalisée par le siège de Novartis (« je vous adresse par ailleurs quelques éléments très synthétiques préparés la semaine dernière par le Global [le siège] sur Lucentis et Avastin », cote 3609).
248. Ce document insiste sur une efficacité garantie pour Lucentis, en comparaison avec les incertitudes s’agissant d’Avastin, soulignant en particulier les risques d’infection liés aux injections intravitréennes et à l’absence d’études cliniques sur les potentiels effets secondaires (« Lucentis is specifically designed and proven safe for use in the human eye (…) No proper data are available on safety or serious side effects with Avastin use in wet AMD (…) its safety as a treatment for wet AMD is unknown », cote 3612).
249. Ce document envisage également de travailler avec des KOL et des associations de patients, afin de diffuser le message auprès des médias européens (cote 6311).
La présentation de mars 2008 « Lucentis vs Avastin total treatment cost »
250. En mars 2008, Novartis a préparé un argumentaire visant à justifier le prix élevé de Lucentis, par rapport au coût d’Avastin (cotes 3588 à 3591).
251. L’objectif de cette présentation était de montrer que, dans certains cas, la différence de coût entre les deux produits n’était pas aussi élevée. Pour chacun des scenarii, la présentation concluait : « Lucentis proven safety and efficacy clearly justifiy selection over Avastin » (traduction libre : « L’efficacité et la sécurité prouvées de Lucentis justifient la sélection de Lucentis de préférence à Avastin »).
252. Le laboratoire a ainsi préparé une « argumentation économique autour de Lucentis », en préparation du congrès de la société française d’ophtalmologie (ou « SFO »), devant se tenir en mai 2008 (cote 3576).
Les documents « préparation du media training / Lucentis »
253. Deux documents de travail relatifs à un « media training », vraisemblablement en vue d’une conférence au Club francophone des spécialistes de la rétine, montrent que Novartis se préparait à répondre à des questions sur la sécurité et l’efficacité d’Avastin (cotes 3593 et 3595, cotes 3604 et 3605).
254. Novartis entendait notamment insister sur les risques pris par les médecins utilisant ce médicament : « Par conséquent, les médecins qui utilisent Avastin en dehors de son indication le font à titre expérimental et engagent leur responsabilité civile en cas de complication » (cote 3593).
Le document « Suivi Business Rétine » du 24 avril 2008
255. Un document, intitulé « Suivi Business Rétine », du 24 avril 2008, montre que les visiteurs médicaux de Novartis ont mis en œuvre sur le terrain des actions auprès des centres utilisant Avastin (cotes 3657 à 3696).
256. Ce document fait en effet état d’un « Plan d'action VM [visiteurs médicaux] Rétine et KAM [‘Key account manager’ en charge des relations avec les services hospitaliers] sur les centres Avastin existants », indiquant « Reste 10 centres identifiés » (cote 45134).
257. Ainsi, Novartis concentrait ses efforts sur les centres d’injection utilisant Avastin afin qu’ils modifient leur pratique, au profit de Lucentis. Cette action semble avoir donné des résultats, puisqu’il est indiqué : « Situation résolue aux 15/20, à la fondation Rothschild, à Grenoble, à Lille, à Toulon. Situation toujours bloquée à Dijon, Nantes, Cannes, Limoges, Hôtel Dieu... le plus souvent pour des raisons financières (ces centres gagnent plus en utilisant l'Avastin plutôt que Lucentis) » (cote 45134). De même, le document indique : « le nombre de centres utilisant l'Avastin a diminué (-8) grâce aux actions conjointes des délégués Rétine et des KAMs [‘Key account manager’ en charge des relations avec les services hospitaliers]. Reste 10 centres Avastin identifiés » (cote 3683).
258. Enfin, ce document fournit des informations sur la structure du réseau de délégués médicaux en ophtalmologie de Novartis (cotes 3691 à 3695). À cette époque, ce réseau se composait de six « visiteurs médicaux Rétine » et de dix-sept « visiteurs médicaux ophtalmologie », qui effectuaient chacun en moyenne 6,6 visites par jour.
Le « 2009 Retina Business Plan » de juillet 2008
259. Dans un document intitulé « 2009 Retina Business Plan » de juillet 2008, Novartis présente Avastin comme une réelle menace : « Avastin threat is concrete (Gefal Study) » (traduction libre : « la menace Avastin est concrète (étude Gefal) », cote 45083) ou encore : « Avastin (…) becoming a potential real threat » (traduction libre : « Avastin (…) devient une potentielle menace réelle », cote 2440).
260. À ce titre, Novartis préconise dans ce document les démarches à suivre pour minimiser l’impact de l’étude GEFAL, qui devait commencer à la fin de l’année 2008 (« Minimize H2H studies (Gefal) impact : Secure Lucentis price, closely monitor the implementation, communicate on insufficiencies », traduction libre : « Minimiser l’impact des études H2H (Gefal) : sécuriser le prix de Lucentis, contrôler de près l’application, communiquer sur les insuffisances », cote 2440). Novartis redoutait en effet que cette étude conduise à l’octroi d’un PTT pour Avastin et, partant à une diminution significative du volume de ventes de Lucentis à l’hôpital (« TTP could lead to a significant decrease of sales volume in the hospital segment », cote 45133) ainsi qu’à la baisse du prix de Lucentis (« Expected impact in 2010 [2011]: Avastin Temporary Therapeutic Protocol (TTP) and Lucentis price decrease », cote 45083).
261. Le document propose donc un plan d’action visant à limiter la part de marché d’Avastin à moins de 10 % (« action plan : (…) contain Avastin < 10 % in wAMD [wet age-related macular degeneration, traduction : DMLA] », cote 2447) ou encore à bloquer son large usage dans les autres indications pour lesquelles Lucentis n’avait pas d’AMM (« our strategy (…) block Avastin broadly usage in other indications (DME [diabetic macular Edema], RVO) », cote 2430). Cette stratégie est répétée à de nombreuses reprises dans le document (cotes 2468 et 2480).
262. Parmi les nombreuses mesures envisagées, Novartis préconisait de développer un discours mettant en cause la pertinence de l’étude GEFAL, en insistant notamment sur les failles méthodologiques et les problèmes non traités dans cette étude, l’objectif étant de centrer la communication sur les insuffisances de l’étude et d’essayer de convaincre que ses résultats seraient publiés trop tardivement (« outline methodology gaps, unaddressed issues, Drive communication on insufficiencies, Try to convince that GEFAL results will come too late », cote 2468).
263. Pour ce faire, le projet de Novartis était d’obtenir le protocole de l’étude GEFAL, de développer un lobbying dirigé vers le Professeur Y…, directeur de l’étude, ainsi que vers les principaux chercheurs impliqués, ou encore de remettre en cause la conception de l’étude et de communiquer sur ses insuffisances (« get the protocol, KOL lobbying (Pr Y… + main investigators, challenge Gefal design and communicate on insufficiencies », cote 2470).
264. En outre, Novartis prévoyait de développer et souligner des éléments de différenciation de Lucentis vis-à-vis d’Avastin sur les questions d’efficacité, de sécurité et de tolérance (« difference on efficacy, safety or tolerability (…) List Avastin side effect to highlight Avastin deficiency », cote 2468).
265. Enfin, la stratégie de Novartis comportait un volet juridique, dont l’objectif était de définir les moyens de contrer les éventuelles initiatives des pouvoirs publics pour « légaliser » l’usage d’Avastin (« Legal strategy to counter Avastin legalization », cote 2468).
266. L’élaboration de ce discours devait être prise en charge par le siège, avant sa diffusion par les équipes françaises (cote 2471), ainsi que l’illustre la reproduction ci-dessous :
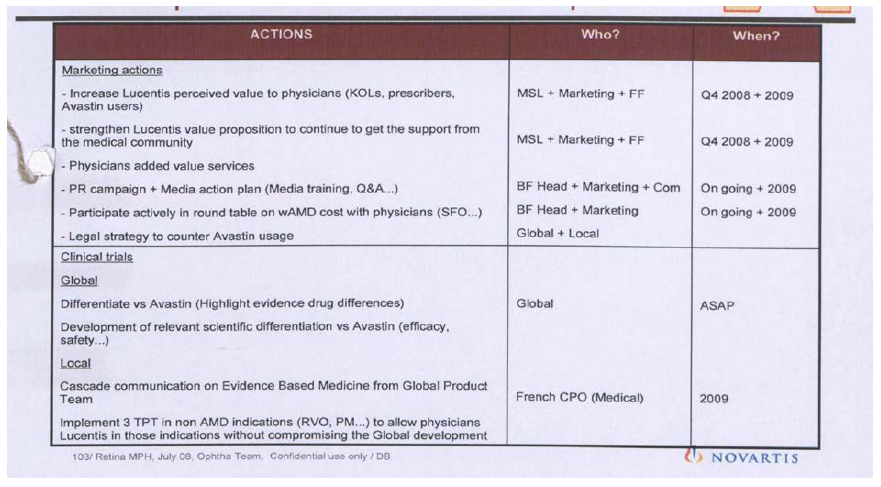
b) Les éléments de communication au cours de l’année 2009
La rencontre du 19 juin 2009 entre Novartis Pharma et le siège du groupe
267. Le 19 juin 2009, une rencontre entre l’équipe du siège de Novartis et celle de Novartis Pharma en charge de Lucentis a été organisée (cotes 2206 et 2207).
268. Le support de présentation de cette rencontre mentionne un plan d’action, auprès des KOL, visant à différencier Avastin de Lucentis, en particulier sur la question de la sécurité, afin d’éviter qu’Avastin ne soit considéré comme un produit concurrent de Lucentis (« Avoid Avastin to be considered as competitor (for KOL). Action plan to differentiate Lucentis from Avastin, including safety », cote 45132).
Le « Lucentis C4 Plan 2009-2014 wAMD [wet age-related macular degeneration]» de juillet 2009
269. Dans un document intitulé « Lucentis C4 Plan 2009-2014 wAMD [wet age-related macular degeneration] », de juillet 2009, Novartis expose ses impératifs stratégiques concernant Lucentis pour les quatre années à venir (cotes 2365 à 2417).
270. Parmi ces objectifs, on retrouve notamment la maximisation du taux de pénétration de Lucentis, en minimisant l’impact d’Avastin, ou encore la réduction de l’utilisation d’Avastin dans les autres indications que la DMLA (« Maximize Lucentis penetration rate and minimize Avastin impact » ; « Reduce Avastin usage in non AMD indications (local TPT prospective studies, Global clinical studies, new Indications) », cote 2366).
271. Le même document note ainsi qu’une des principales faiblesses de Lucentis est l’absence de différenciation prouvée cliniquement et la perception de son prix comme élevé (« Key issues (…) No proven clinical differentiation between Lucentis & Avastin, and Lucentis price perceived as high », cote 2376). Novartis entend, par conséquent, se préparer au cas où l’étude GEFAL démontrerait la non-infériorité d’Avastin par rapport à Lucentis en termes d’efficacité, afin d’en minimiser l’impact (« GEFAL study has started (…). We need to be prepared in case of proven non-inferiority in terms of efficacy to minimize impact », cote 2377).
272. En particulier, Novartis notait que les études cliniques comparatives allaient probablement montrer la non-infériorité d’Avastin. Il relevait par ailleurs qu’une majorité de médecins français anticipaient que les études de comparaison en cours – et notamment GEFAL – allaient démontrer l’équivalence d’Avastin et de Lucentis, non seulement en termes d’efficacité, mais également de sécurité, et que des actions pour différencier Lucentis par rapport à Avastin devaient être rapidement mises en place (« Head to Head trials will likely show non inferiority. Actions to differenciate Lucentis vs Avastin should be quickly set up as physicians already anticipate similar safety and efficacy (56 %) », traduction libre : « Les études de comparaison vont probablement démontrer la non infériorité. Des actions pour différencier Lucentis d’Avastin devraient rapidement être mises en place, puisque les physiciens anticipent déjà une sécurité et une efficacité similaire (5 6%) », cote 2392). Les équipes de Novartis devaient donc se préparer à utiliser les arguments développés par le siège sur les effets secondaires d’Avastin, en réponse aux arguments des médecins qui seraient convaincus de la supériorité d’Avastin (« MSL communication on Roche DOL about Avastin side effects, not proactively but in defense if physicians are convinced about Avastin superiority », cote 2392).
273. Ce document contient en outre des éléments sur les actions de Novartis auprès des centres d’injection. Ainsi, lorsqu’un médecin d’un de ces centres déclarait utiliser Avastin, la stratégie à mettre en place consistait à l’inviter à une réunion régionale et à lui présenter les insuffisances de l’étude GEFAL, ou encore, à développer un argumentaire sur les effets secondaires d’Avastin (« invitation to a regional meeting (…), presentation of GEFAL gaps, report of Avastin side effects », cote 2407).
c) Les éléments de communication au cours de l’année 2010
Le guide ACUITY pour l’année 2010
274. Dans le cadre du programme ACUITY, – défini par Novartis en réponse à une demande d’information des services d’instruction comme suit : « ACUITY (‘Advancing Collaboration and Understanding In OphThalmologY’) était le nom générique utilisé par Novartis pour désigner les Comités consultatifs sur la rétine composés de 15 à 20 ophtalmologues reconnus par leurs pairs comme des leaders dans ce domaine » (cote 45136) – le siège de Novartis a transmis aux filiales nationales un ensemble de documents destinés à permettre aux équipes du groupe d’encadrer les débats au sein des comités consultatifs DMLA.
275. Parmi ces documents, se trouvait un guide de modération et de discussion relatif aux études de comparaisons (H2H) entre Avastin et Lucentis (cotes 14937 à 14955), accompagné d’une présentation intitulée « Head-to-head trials of Lucentis versus Avastin » (cotes 14957 à 14977).
276. Ces deux documents contenaient de nombreux éléments de critique de la méthodologie des études CATT et IVAN, tant sur la question de l’efficacité comparée d’Avastin et Lucentis, que sur celles de la tolérance et de la sécurité. En particulier, Novartis affirmait que le protocole de l’étude CATT permettait au bevacizumab (Avastin) d’être 30 % moins efficace que le ranibizumab (Lucentis) et d’être pourtant considéré comme non-inférieur (« CATT : wide non-inferiority margin allows to be 30 % less efficacious », cote 14974).
277. En conclusion, Novartis affirmait que les études CATT et IVAN ne permettraient pas d’obtenir des conclusions sérieuses tant sur l’efficacité que la sécurité d’Avastin en ophtalmologie (« it is unlikely that any of the current ongoing head-to-head trials are powered for safety > larger studies powered to detect potentially significant systemic adverse events may be required. Possible Biais for non-inferiority (…) > Limited scientific value and interpretability », traduction libre : « Il est peu probable que les études en cours soient probantes pour la sécurité > de plus larges études réalisées pour détecter les éventuels événements indésirables systémiques graves seront probablement requises. Il y a un biais probable sur la non-infériorité (…) > valeur scientifique et interprétation limitée », cote 14976).
Le document « Lucentis Defense Workshop »
278. Un document interne de Novartis, intitulé « Lucentis Defense Workshop » diffusé dans le cadre d’un atelier organisé en 2010, illustre l’objectif à suivre par Novartis pour se défendre des attaques répétées sur Avastin : il s’agira pour l’entreprise de créer un environnement favorable au prix de Lucentis et à son remboursement postérieurement aux études de comparaison (qui seront publiées entre 2011 et 2014) (« Goal is to create an environment that is supportive of Lucentis price & reimbursment post-H2H », cote 4356).
279. Parmi les actions envisagées, Novartis projetait de développer les relations avec les professionnels qui étaient favorables à Lucentis, en particulier par le biais d’un plan d’action vis-à-vis des KOL, notamment au travers du programme ACUITY, et de certains conseils consultatifs régionaux (« develop advocates who recognise the Lucentis value proposition. Develop PA-KOL advocacy plan, maximise ACUITY, hold local advisory boards », cote 4358), ou encore d’augmenter l’incertitude autour d’Avastin, en soulignant a contrario les certitudes sur l’innocuité de Lucentis et en communiquant sur les limites des études de comparaison (« increase uncertainty around Avastin use (…), emphasise certainty of Lucentis safety (…), communication of H2H safety deficiencies », cote 4358).
La présentation « Lucentis advocacy plan » du 25 mai 2010
280. Dans la présentation « Lucentis advocacy plan » du 25 mai 2010, le siège de Novartis revient sur les différences pharmacocinétiques entre Avastin et Lucentis, sur le caractère jugé critiquable de la méthodologie des études CATT et GEFAL, ainsi que sur les règles relatives à la responsabilité médicale en cas de prescription « hors AMM » (cotes 13819 à 13940).
281. La présentation contient par ailleurs une section dédiée aux éléments scientifiques alors disponibles sur les risques liés à l’utilisation d’Avastin en ophtalmologie. En conclusion, elle souligne que des études supplémentaires seraient nécessaires pour déterminer si les risques éventuels envisagés sont une cause sérieuse d’inquiétude pour Avastin (« Larger scale and longer follow-up required to determine whether these events are a safety concern with bevacizumab [Avastin] », traduction libre : « Un suivi à plus grande échelle et plus long est nécessaire pour déterminer si ces événements sont une préoccupation de sécurité avec le bevacizumab [Avastin] », cote 13880).
282. La présentation contient en outre des développements sur une série de cas de réaction inflammatoire intraoculaire sévère intervenus au Canada, à la suite de l’utilisation d’Avastin en injection intravitréenne et ayant fait l’objet d’une communication publique de Roche (cote 13866). Le document précise d’ailleurs que ces cas sont examinés de près par Genentech et Roche, qui travaillent sur un plan d’action sur ce sujet et tiennent Novartis informé (« Next steps. Genentech is working with Roche to uncover the reasons of this cluster of TASS [toxic anterior segment syndrome] and Roche/Genentech action plan. (…) Novartis is being kept informed », traduction libre : « Prochaines étapes. Genentech travaille avec Roche pour déterminer les raisons de ce cluster de syndrome toxique du segment antérieur et un plan d’action Roche/Genentech », cote 13867).
La réunion « Advocacy Lucentis » du 26 mai 2010
283. Le 26 mai 2010, a eu lieu au sein de Novartis une réunion, réunissant l’ensemble des personnes impliquées dans la promotion de Lucentis : le Président directeur général (M. Z…), les équipes du marketing « Market Access », la Franchise Ophtalmologie, regroupant les forces commerciales présentes en région et spécialisées sur les médicaments d’ophtalmologie, les « Key Account Managers » (ou « KAM »), la direction juridique, ou encore la direction des affaires pharmaceutiques (cote 2126).
284. Le document de support de cette réunion (cotes 2125 à 2141) précise en introduction que son objectif est de « faire face aux études Avastin et optimiser le lancement dans la DME [diabetic macular Edema]» (cote 2125).
285. Ainsi, l’un des objectifs de la journée était de construire un plan de communication au niveau local, permettant de soutenir Lucentis et de mettre en avant les défauts d’Avastin dans le traitement de la DMLA, compte tenu notamment des dernières études scientifiques (« Optimal preparation at local level maximies opportunity to secure current sales and future growth due to the following events : results of the H2H trials CATT (US), IVAN (UK) (…) Robust advocacy plan to build support for Lucentis and highlight deficiencies of Avastin in wAMD », traduction libre : « Une préparation optimale au niveau local maximise l’opportunité de sécuriser les ventes actuelles et la croissance future du fait des événements suivants : résultats des études H2H CATT (US), IVAN (GB) (…) un plan robuste de promotion est à construire pour soutenir Lucentis et metre en avant les insuffisances d’Avastin pour la DMLA », cote 2127).
286. La journée a commencé avec une présentation de la stratégie du groupe Novartis, reprenant notamment les éléments déjà définis en 2009 en termes de lutte contre la présence d’Avastin en ophtalmologie (cote 2131). Ensuite, trois ateliers successifs ont été proposés aux participants, afin de s’approprier la stratégie de Lucentis, développer des réponses à objections et des messages clés, et définir des plans d’action en région (cote 2134).
287. S’agissant plus particulièrement des messages clés et réponses à objections, les présentations insistent à nouveau sur les effets indésirables systémiques « potentiellement plus importants » pour Avastin. Ils évoquent également les problèmes intervenus aux États-Unis et au Canada, liés au reconditionnement d’Avastin (cote 2137). Elles affirment à nouveau que les études de comparaison en cours, telles que GEFAL et CATT, sont insuffisantes d’un point de vue méthodologique (« Méthodologie des études Avastin H2H vs Lucentis est challengeable ») et indiquent que « le médecin peut engager sa responsabilité civile, pénale, administrative et disciplinaire si la prescription hors AMM d'un produit n'est pas justifiée » (cote 2138).
288. Ces messages ont ensuite été utilisés pour développer des réponses à objections utilisées par les forces commerciales de Novartis dans leurs contacts avec les professionnels, dans le cadre de plans d’action en région (cotes 2140 et 2141).
La réunion « M3PH » de juin 2010
289. En juin 2010, une réunion dédiée à la stratégie concernant Lucentis pour l’année 2011 a eu lieu au siège de Novartis (cotes 13942 à 14031).
290. Concernant Avastin, le document de présentation indique qu’à cette époque, l’usage d’Avastin était désormais limité à quatre principaux centres d’injection : l’Hôtel-Dieu à Paris, les Hospices civils de Lyon, la clinique Sourdille à Nantes et l’hôpital clinique Claude Bernard à Metz (« Avastin usage in wAMD now limited to four main centers in France », cote 13977). De manière générale, les hôpitaux publics constituaient, à l’époque, la cause principale d’inquiétude s’agissant de l’emploi d’Avastin, Novartis estimant que ces derniers étaient plus « vulnérables » à la pénétration d’Avastin que les médecins de ville (« Public hospitals are more vulnerable to Avastin penetration than office based practice », cote 13978).
291. Dans ce contexte, Novartis anticipait que les études de comparaison en cours – notamment CATT et GEFAL – allaient conclure à l’équivalence entre Avastin et Lucentis en termes d’efficacité (« No proven clinical differentiation of Lucentis vs. Avastin. H2Hs likely to demonstrate non-inferiority on efficacy », cotes 13979) et impacteraient les choix tant des autorités de santé que des médecins individuels (« Avastin H2H studies results will impact both HA [health authorities] and medical community », cote 13980). Le document établit ainsi une liste des menaces potentielles pouvant découler de la publication de ces études. En premier lieu, Novartis considère que, avec une probabilité estimée à 90 %, les organismes payeurs pourraient utiliser Avastin comme comparateur pour faire baisser le prix de Lucentis (« payors use the trial results to pressure NVS [Novartis] to reduce price. H2H positive result will legitimate Avastin as price comparator at time of DME [diabetic macular Edema] negotiation /wAMD re-listing negotiation », cote 13980). En deuxième lieu, Novartis estime, avec une probabilité estimée à 60 %, que les autorités françaises pourraient instaurer un PTT pour Avastin à l’hôpital, tout en soulignant néanmoins la possibilité de contester une telle mesure (« The trial will enable authorities to allow restricted Avastin use in public hospital through temporary therapeutic protocol (ATU like) process (…) PTT authorization could be challenged as alternaites are available for wAMD », cote 13980). En dernier lieu, Novartis estime que les médecins seront influencés par les résultats des études et augmenteront leur utilisation d’Avastin, tout particulièrement dans les hôpitaux (« physicians will be influenced by trial results and will increase their use of Avastin (particularly in hospital practices) », cote 13980) :
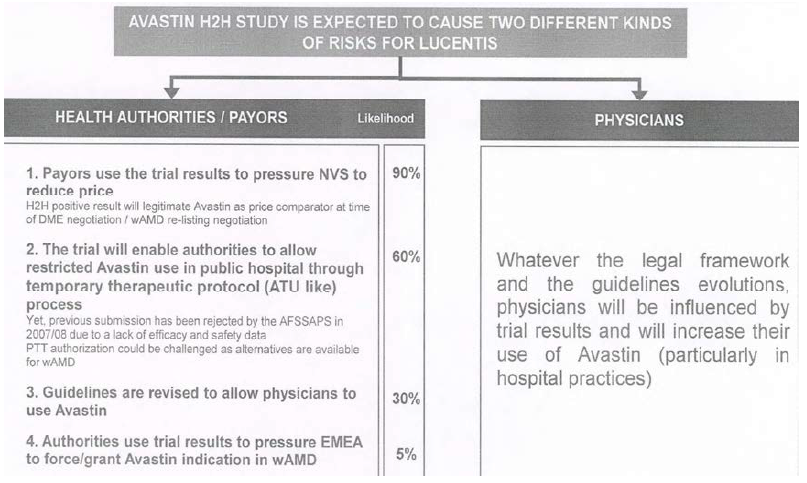
292. Le document prévoit la mise en place d’un vaste plan de communication vis-à-vis tant des autorités que des professionnels, afin de limiter l’utilisation d’Avastin. Ce plan inclut notamment le financement de nouvelles études, de services innovants, et la fourniture de produits gratuits (« > Project funding or mananagement for leaders > Clinical studies, > (…) Sampling » cote 13983). Il implique également la diffusion de « messages clés » insistant sur les différences en termes de pharmacologie, d’efficacité et de sécurité entre Lucentis et Avastin (cotes 13989 et 13991).
293. Les principaux éléments de message sont contenus dans un « Elevator speech » (« argumentaire éclair ») qui reprend sous une forme synthétique les « messages clés », et insiste sur l’incertitude scientifique et réglementaire entourant l’utilisation d’Avastin en ophtalmologie, rapprochée, a contrario, des certitudes sur l’innocuité de Lucentis (« Avastin is different from Lucentis (…) Avastin is uncertain (…) Avastin is unapproved (…) NVS [Novartis] is a true partner for ophtalmology », traduction libre : « Avastin est différent de Lucentis (…) Avastin est incertain (…) Avastin n’est pas autorisé (…) NVS [Novartis] est un vrai partenaire pour l’ophtalmologie », cote 13988). Le document revient également sur les limites méthodologiques des études disponibles (« la méthodologie des études Avastin H2H vs Lucentis est challengeable [peut être contestée] », cote 13991) et sur le risque juridique encouru lorsqu’un médecin décide d’utiliser Avastin (« Le médecin et le pharmacien engagent leur responsabilité », cote 13991).
294. Le document de présentation indique ainsi que les différents supports et argumentaires rédigés par Novartis devaient être diffusés progressivement au sein du groupe, notamment à l’ensemble des forces commerciales lors du séminaire prévu entre le 13 et le 17 septembre 2010 (cote 13992).
295. L’argumentaire devait ensuite être diffusé à un ensemble de KOL, en fonction de leur influence respective telle que définie par la pyramide (« Avastin / Lucentis advocacy communication is to be rolled out accordingly to RS influence pyramid », cote 13993). À cet égard, la présentation note que le développement de projets collaboratifs avec les KOL, ainsi que l’accroissement du nombre de rencontres en face à face, ont permis une meilleure diffusion du discours depuis un an (« For 1 year, collaborations and relations with KOL have been greatly developped allowing a straight communication on Avastin/Lucentis issues », traduction libre : « Depuis 1 an, les collaborations et relations avec les KOL ont été fortement développées, permettant une communication directe sur les questions Avastin/Lucentis », cote 13993).
296. Enfin, Novartis prévoit un certain nombre d’actions plus ciblées localement, notamment vis-à-vis de certains centres d’injection, appelés « key centers » (cotes 13994 à 13997).
297. L’un des centres particulièrement visés par Novartis est celui des Hospices civils de Lyon, et notamment le Professeur Y…, qui dirige l’étude GEFAL (cf. paragraphes 119 à 124 ci-dessus). Dans ce contexte, Novartis prévoit de « maintenir occupé » l’intéressé, notamment en l’invitant à ses propres comités consultatifs ou en favorisant son implication dans d’autres projets en Rhône-Alpes Auvergne (« Key lever : Keep Y… busy », traduction libre : « Un levier essentiel : maintenir Y… occupé » cote 13996).
Les réunions « LBT » (ou « Local Brand Team ») de juillet et septembre 2010
298. Le 1er juillet 2010, une réunion « LBT » (ou « Local Brand Team ») s’est tenue au sein de Novartis (cotes 4628 à 4630).
299. Le compte rendu de cette réunion montre qu’à cette occasion, Novartis a décidé de formaliser un document de synthèse d’« objections/réfutations » sur le sujet de l’utilisation « hors AMM » d’Avastin et d’organiser une formation des équipes commerciales sur ce point (« partage sur le diagnostic d’un besoin de formalisation d’un document de synthèse de type ‘objections/réfutations’ à destination d’un usage interne Novartis (…) Organisation post juillet d’une session de formation/training avec l’ensemble des KAM [‘Key account manager’ en charge des relations avec les services hospitaliers] sur les objections/réfutations Avastin (…) Test du document à envisager avec le board DMLA (en groupe ou individuel) », cote 4628).
300. Ce document montre également que Novartis a décidé de préparer une présentation type Powerpoint portant sur les différences entre Avastin et Lucentis, aux fins de présentation aux KOL (« Kit de slide MSL pour présentation KOL et axé sur les différences entre Avastin et Lucentis (pharmacologie, safety) », cote 4629). Novartis a également décidé de constituer un groupe de travail sur la « responsabilité juridique et pharmaceutique » des praticiens utilisant Avastin (cote 4629).
301. Enfin, le compte rendu de réunion indique que les représentants de Novartis « pourront avoir une action plus proactive, mais coordonnée et ciblée sur centres utilisateurs ou proches de basculer ou sur les centres faisant partie du halo d’influence des autorités de santé (cote 4629).
302. Cette répartition des rôles et des types de discours a été affinée dans le cadre d’une autre réunion « LBT » du 22 septembre 2010 (cotes 14850 à 14854). Le compte rendu de cette réunion montre que le document « objections / réfutations » devait être utilisé uniquement en interne au siège, à l’exception du résumé opérationnel de l’« Elevator Speech », qui devait être remis aux forces commerciales au cours des sessions de formation, prévues en septembre, décembre 2010 et janvier 2011 (« Document ‘Objections/réfutations’ utilisation en interne au siège. Pas de diffusion terrain à l’exception de l’executive summary ‘Elevator Speech’ (…) Document ‘Elevator Speech’ remis pendant le séminaire de septembre + formation. Communication et formation prévue pour les KAM [« Key account manager » en charge des relations avec les services hospitaliers] en décembre 2010 / janvier 2011», cote 14850).
303. S’agissant des « slides » sur les différences de sécurité entre Avastin et Lucentis, ces éléments devaient être présentés au comité consultatif DMLA de Novartis le 17 octobre 2010 (« présentation du slide kit au board DMLA du 17/10 pendant l’AAO [American Academy of Ophtalmology] », cote 14850).
La réunion de préparation du « séminaire ophta » du 1er juillet 2010
304. Le 1er juillet 2010, une réunion de préparation du séminaire des forces commerciales de la franchise ophtalmologie de Novartis a eu lieu (cotes 15685, 15687 à 15688 et 45111).
305. Le compte rendu de cette réunion indique qu’une session de formation, intitulée « Training Avastin », était prévue pendant une demi-journée. Cet exercice consistait à combiner l’ensemble des « objections » des médecins sur le sujet et de fournir aux délégués médicaux des arguments en réponse. Par ailleurs, pendant tout le séminaire, des agents de l’agence de communication et d’organisation d’événements engagée pour organiser le séminaire pouvaient aborder les délégués hospitaliers et les chargés de relation clientèle pour les « tester » sur les réponses à donner à ces objections. Les employés de l’agence PLB devaient donc également être formés sur le sujet (cotes 45111 et 15687, « Des animateurs de l’agence PLB pourront à n’importe quel moment de la semaine, aborder les DHs / CRCs pour les ‘tester’ sur les réponses à objection. Il est donc nécessaire de briefer l’agence sur les questions/objections à poser et sur les arguments »).
La présentation du 28 septembre 2010
306. Une présentation du 28 septembre 2010, intitulée « Addressing the challenge of unlicensed Avastin and H2H data release » (traduction libre : « répondre au défi de l’usage d’Avastin hors AMM et de la sortie des études de comparaison »), contient à nouveau un plan complet d’application de la stratégie vis-à-vis d’Avastin et de diffusion des messages définis en interne à l’ensemble des parties prenantes du secteur de la santé (cote 14038 à 14054).
307. Plus spécifiquement, ce document prévoit notamment que les référents médicaux (« MSL ») devaient contacter les cinquante KOL les plus influents et leur fournir les « messages clefs » de Novartis sur Avastin. Ces messages devaient également être diffusés aux médecins les plus orientés vers Avastin (« selected Avastin lovers », traduction libre : « amoureux d’Avastin selectionnés », cote 14045). La même « slide » souligne que ces messages devaient être relayés de manière très prudente aux associations de patients et à d’autres influenceurs (« Very careful awareness to patient associations leaders and/or influencers from MSL », cote 14045). Enfin, le document précise que les autorités de santé et organismes payeurs devaient être destinataires de l’« Elevator Speech » de Novartis.
308. Ce document contient également une cartographie complète des KOL présents dans les comités consultatifs de Novartis, en fonction de leur capacité à répercuter l’argumentaire de l’entreprise (cotes 14046 à 14050) :

L’argumentaire des délégués médicaux de septembre 2010
309. Un document intitulé « Training Lucentis vs Avastin Région 93 », communiqué au cours d’un séminaire en ophtalmologie organisé en septembre 2010, reprend les « arguments clés Lucentis vs Avastin » (cotes 3472 et 3473). Si ce document précise être à usage interne uniquement, et comme ne devant « en aucun cas » être remis aux médecins (« ce document est réservé uniquement à l’usage des délégués médicaux et ne peut en aucun cas être remis au corps médical », cote 3473), il comporte néanmoins le discours devant être délivré oralement par les délégués médicaux.
310. Ces « arguments clés » correspondent à ceux contenus dans l’« Elevator speech » susvisé, tendant à insister sur l’incertitude scientifique et réglementaire entourant l’utilisation d’Avastin en ophtalmologie, rapprochée, a contrario, des certitudes sur l’innocuité de Lucentis (« Avastin est différent de Lucentis (…) La tolérance d’Avastin est incertain en ophtalmologie (…) Avastin n’est pas approuvé dans la DMLA (…) Novartis est un partenaire à vos côtés en ophtalmologie », cote 3473).
311. L’argumentaire repose sur des affirmations conditionnelles s’agissant des effets indésirables (« EI [effets indésirables] probablement très sous-estimés (utilisation hors AMM) (…) Existences de spécificités d’Avastin qui pourraient expliquer certains EI [effets indésirables] constatés », cote 3473). Il souligne également le caractère insuffisant des études de comparaison sur la sécurité d’utilisation d’Avastin pour justifier l’obtention d’une AMM pour ce produit (« les études H2H en cours sont insuffisantes pour l’obtention d’une AMM ; en particulier car elles n’apporteront pas d’évaluation solide de la sécurité d’utilisation d’Avastin en ophtalmologie », cote 3473) et, enfin, revient sur la responsabilité médicale en cas d’utilisation d’Avastin (« la responsabilité du médecin peut être engagée en cas de problème », cote 3473).
d) Les éléments de communication au cours de l’année 2011
Le « Lucentis Defend plan » d’avril 2011
312. Une présentation d’avril 2011, intitulée « Lucentis Defend vs Avastin – Lucentis Defend Plan », reprend les éléments de la stratégie définie en septembre 2010 (cotes 14092 à 14123).
313. Cette présentation apporte en outre quelques précisions, notamment sur le comportement attendu des MSL du groupe lors de séances du comité consultatif et de contacts bilatéraux avec des KOL. Il leur est ainsi demandé d’insister particulièrement sur les questions de sécurité, en particulier sur les risques d’effets indésirables systémiques liés à l’utilisation d’Avastin, en utilisant les résultats de l’étude Curtis (« MSL communication. Safety focus (…) Serum concentration differences could suggest systemic AE [adverse effects]. CV Safety data from Curtis et Al. », cote 14105). Novartis prévoyait de diffuser très largement les résultats de cette étude, notamment au travers de certains KOL déjà convaincus, en particulier lors de comités consultatifs (« Curtis et al. publication will be leveraged through a dedicated action plan », traduction libre : « La publication de l’étude Curtis sera utilisée comme levier à travers un plan d’action spécifique » cote 14106).
314. Enfin, la présentation insiste plus fortement encore sur la nécessité de rappeler les risques juridiques en termes de responsabilité médicale pour les professionnels utilisant Avastin pour le traitement de la DMLA, en évoquant notamment le précédent du Mediator (« Legal issues and physician liabilities regarding off label prescriptions is also to be highlighted in a Mediator crisis context », cote 14110).
Le « plan d’action à l’approche de la parution de l’étude CATT »
315. Dans une présentation, non datée, intitulée « Avastin Vs Lucentis - Plan d’action à l’approche de la parution de l’étude CATT », Novartis revient sur la stratégie de communication à adopter dans le contexte de la parution imminente des résultats de l’étude CATT (cotes 3104 à 3115).
316. Le document indique que Novartis devait recevoir les résultats de l’étude CATT le 22 avril 2011, soit peu avant le congrès de l’ARVO qui devait se tenir le 1er mai de la même année (cote 3105). Novartis devait donc profiter de ce délai pour former l’ensemble de ses équipes, y compris celles actives en région, et être prête pour diffuser son message aux praticiens, et notamment aux 50 principaux KOL, dès la parution de l’étude (cotes 3106 et 3107).
317. Concernant le message devant être diffusé, celui-ci demeure centré autour des idées clefs de l’« Elevator speech » (ou « discours d’ascenseur »), adapté en fonction des éléments récents, en recentrant le discours sur le débat sur la sécurité. Plus spécifiquement, l’un des messages devant être diffusé est le suivant : « CATT ne prouvera rien en termes de safety : les effectifs insuffisants de CATT ne permettent pas de relever de différences significatives sur le risque de survenue d’effets indésirables rares, comme les événements cardiovasculaires par exemple, entre ranibizumab [Lucentis] et bavacizumab [Avastin].
Ainsi une augmentation du risque d’accidents thromboemboliques due au bevacizumab [Avastin], qui aurait des conséquences dans la ‘vraie vie’, ne peut être mis en évidence dans CATT » (cote 3109). Le document revient également sur la question de la responsabilité du praticien, en cas de prescription « hors AMM » (« Le traitement hors-AMM : quelle est la responsabilité du praticien ? Responsabilité disciplinaire (…) Responsabilité civile (…) Responsabilité pénale », cote 3113).
318. Enfin, la présentation indique que « les études clés de notre stratégie de défense » portent notamment sur l’« Evaluation du taux plasmatique de VEGF avant et après injection de bevacizumab [Avastin] » et sur l’« Evaluation et comparaison du taux plasmatique de VEGF après injections de ranibizumab [Lucentis] et bevacizumab[Avastin] », le laboratoire centrant donc son argumentaire sur le lien entre la concentration en VEGF dans le sang et la survenue d’effets indésirables systémiques (cote 3112).
L’argumentaire économique sur le coût de Lucentis et d’Avastin
319. Dans un courriel du 11 mai 2011, la responsable de la tarification et de l’économie de la santé de Novartis indique que les KAM, « Key account manager » en charge des relations avec les services hospitaliers, ont reçu des éléments de discours sur le coût respectif de Lucentis et d’Avastin (cotes 4209 et 4210).
320. Ces éléments concernaient notamment une évaluation du coût des événements indésirables graves qui seraient causés par Avastin. L’auteur du courriel souligne qu’il n’est pas possible d’estimer, même de façon imprécise, le coût des éventuels problèmes de sécurité liés à l’utilisation d’Avastin en ophtalmologie (« Pour ce qui est des coûts de la safety d’Avastin, pour le moment on ne peut pas faire d’estimation, même ‘louchométrique’ », cote 4209). Pourtant, elle propose des estimations, correspondant à des « ordres de grandeur » des coûts d’hospitalisation.
Le « plan tactique régional Market Access » pour l’Ile-de-France
321. Une présentation, intitulée « plan tactique régional Market Access », développant la stratégie de Novartis pour la région Ile-de-France, le 2 novembre 2011, revient sur les relations avec les équipes de l’un des « centres clés », l’Hôtel-Dieu (cote 15691).
322. Le document indique que cet hôpital utilise Avastin pour les patients hospitalisés et en consultations internes (« utilisation d’Avastin pour patients atteints de DMLA en intra + consult externes (HDJ) », cote 15691). Il rappelle que, le 9 mai 2011, le chef de service de pharmacie de cet établissement a pris publiquement position contre Lucentis et que, le 8 juillet 2011, l’Assistance Publique – Hôpitaux de Paris (« AP-HP ») a pris position en faveur de l’utilisation d’Avastin (« 9 mai 2011 : Pr A… (chef de service pharmacie Hôtel-Dieu et Président de l’académie française de Pharmacie) prend position contre Lucentis dans l’affaire du Figaro. 11 juillet 2011 : Réunion APHP sur l’intérêt de Lucentis vs Avastin (Juste Prescription) conclusions en faveur de l’utilisation d’Avastin dans la DMLA, cote 15691).
323. Concernant cette réunion de l’AP-HP, un verbatim d’une réunion de la commission d’AMM de l’AFSSAPS du 13 octobre 2011, précise que le Docteur B…, vice-président de la commission d’AMM, a indiqué : « au niveau de la Commission du Médicament et des Dispositifs Médicaux Stériles (COMEDIMS) qui a été saisie par ce problème au niveau de l’Assistance Publique - Hôpitaux de Paris (AP-HP), on a considéré (parce qu’on a un autre niveau de décision, d’autres missions) que les pharmacies qui reconditionnaient de l’Avastin et qui par exemple pour l’Hôtel-Dieu faisaient deux millions d’euros d’économies par an et qui considéraient qu’ils avaient le moyen de le faire correctement, (comme ils ont les moyens de faire des préparations de nutrition parentérale ou des chimiothérapies), pouvaient préparer des mono-doses d’Avastin pour injection oculaire »(28).
324. En réaction à cet ensemble d’éléments, la stratégie prévue par Novartis est la suivante : « Mise en place d’une stratégie d’isolement via MSL/DRRIE pour limiter l’impact au-delà de l’établissement » (cote 15691).
Le plan tactique régional Market Access pour la Bourgogne, Franche-Comté, L’Ile de la Réunion et Mayotte
325. Dans une présentation, intitulée « plan tactique régional Market Access », développant la stratégie de Novartis pour la Bourgogne, la Franche-Comté, l’Ile de la Réunion et Mayotte, le 3 novembre 2011, il est indiqué que le CHU de Besançon continuait à l’époque d’avoir recours à des prescriptions « hors AMM » en concurrençant Lucentis.
326. Le document indique que le résultat obtenu à cette époque était le maintien de Lucentis à l’hôpital dans l’attente des résultats de l’étude GEFAL (cote 15694).
e) Les éléments de communication au cours de l’année 2012
La réunion des équipes européennes de Novartis du 24 janvier 2012
327. Le 24 janvier 2012, une réunion des équipes européennes de Novartis a eu lieu (cote 1559).
328. La présentation relative à cette réunion montre que, pour le marché français, le groupe Novartis s’inquiétait de l’impact de l’étude GEFAL sur la renégociation du prix de Lucentis prévue pour la fin 2012 (« Local H2H study (GEFAL data release Q2/Q3) : impact on 5-year price review in Q2/Q3 », cote 45064).
329. Cette présentation inclut ainsi un plan de réponse à la publication des résultats à un an de l’étude IVAN (« Novartis post-CATT reactions / IVAN plans (…) Novartis preparation are underway to develop internal/external IVAN study communications materials to ensure rapid response to the 1-yr [year] release », traduction libre : « Réactions de Novartis après l’étude CATT / IVAN (…) Préparation de Novartis en cours pour développer des supports de communication internes/externes sur l’étude IVAN, pour garantir une réponse rapide à la publication des résultats à un an », cote 1571). Ce plan incluait des discussions avec les KOL, les autorités de santé et les associations de patients (« IVAN / CATT 2 2y [year] communication plan will be similar to CATT 1y [year] with rapid communication to all stakeholders (…) Physicians/KOLs (…) Patient groups (…) Regulatory EU comm. [European Commission] (…) », traduction libre : « Le plan de communication pour IVAN/CATT 2 ans sera similaire à celui de CATT 1 an, avec une communication rapide à toutes les parties prenantes (…) Physiciens/KOL (…) Associations de patients (…) Reglementaire Commission européenne », cote 1572). Pour cela, Novartis prévoyait de mettre à jour son argumentaire (« Summary of Lucentis and Avastin high level messages », traduction libre : « Résumé des messages de haut niveau sur Lucentis et Avastin », cote 1572), dont la structure demeurait la même, focalisé sur la potentialité d’effets indésirables plus importants avec Avastin (« Safety concerns : there is an emerging body of evidence indicating that the risk of ATEs [adverse effects] may be higher with unlicensed Avastin compared to Lucentis », traduction libre : « Préoccupations de sécurité : il y a un ensemble de preuves qui fait surface indiquant que les risques d’événements indésirables pourraient être supérieurs avec l’utilisation ‘hors AMM’ d’Avastin en comparaison avec Lucentis », cote 1572) et la critique de la pertinence statistique des études de comparaison en cours (« H2H not powered for safety ; particularly for the rates of rare and life-threatening events », traduction libre : « Les études H2H [de comparaison] ne sont pas dimensionnées pour la sécurité ; en particulier pour les taux d’événements rares et mortels », cote 1572), en comparaison avec les garanties de Lucentis (« Lucentis Licensed (…)
Designed for the eye (…) Lucentis has well-characterized safety profile », traduction libre : « Lucentis est autorisé (…) Créé pour un usage dans l’œil (…) Lucentis a un profile de sécurité bien établi », cote 1572).
330. Outre la réaction prévue aux études de comparaison, la présentation montre que Novartis entendait faire usage dans sa communication du probable futur changement par Roche du RCP d’Avastin (« leverage likely upcoming label change of Avastin in accounts / regions at risk », cote 1566). Sur ce point, une présentation spécifique datée de janvier 2012 – mise à jour en février 2012 – détaille la stratégie de Novartis pour exploiter cette évolution dans sa communication concernant l’usage d’Avastin en ophtalmologie (cotes 13764 à 13767). Elle souligne en particulier que si Novartis ne développe aucune communication proactive sur le sujet, cela risque de demeurer un « non-événement » (« if no proactive information from Novartis this will be a non-event… », cote 13766). A contrario, une communication trop large pourrait être contre-productive (« …but a large communication campaign has a high risk to be counter productive for Novartis… », cote 13766). En conséquence, les équipes en charge des relations avec les autorités de santé recommandent une communication ciblée, limitée aux comités consultatifs et à des entretiens bilatéraux, mise en œuvre par les MSL du groupe (« …so regulatory workstream team recommend a targeted communication to key stakeholders. Advisory board. Face to face meetings. MSLs (Defend vs. Avastin slides) », cote 13766).
La conférence téléphonique du 31 janvier 2012 et les « messages clés » concernant Lucentis/Avastin
331. Le 31 janvier 2012, une conférence téléphonique regroupant des personnes de l’ensemble du groupe Novartis impliquées dans la commercialisation de Lucentis a eu lieu (cote 1554). L’un des thèmes de cette conférence téléphonique était la lutte contre l’usage « hors AMM » d’Avastin en ophtalmologie (« Minimize the unlicensed use of Avastin in the eye », cote 1557).
332. À cette époque, le groupe Novartis continuait de centrer son action contre l’usage d’Avastin en ophtalmologie, en insistant sur les questions de sécurité. Il était ainsi notamment prévu de favoriser un plus grand nombre de publications soulevant le « sujet des préoccupations de sécurité » (« achieve higher number of publications raising ‘safety concern story’ », cote 1557), de développer un plan de communication pour la publication des résultats à deux ans de l’étude CATT (« develop a communication plan for the wave 2 H2H data (IVAN, CATT2, etc.) based on the learnings of the CATT 1 roll-out », cote 1557) ou encore de rédiger un document d’une page, à destination des autorités de santé (« develop 1 pager statement focusing on Avastin safety issues to patients vs Lucentis certainty and value », cote 1557).
333. Ce dernier document, intitulé « Lucentis/Avastin - Messages clés », a été rédigé en reprenant les messages clés déjà présents dans la stratégie élaborée par Novartis les années précédentes. Ce texte affirme notamment que « les études [de comparaison] en cours ou à venir ne sont pas suffisantes pour l’obtention d’une AMM », que « des études rétrospectives publiées (Curtis, Gower) font état de signaux sur la tolérance d’Avastin », ou encore que « les résultats de l'étude CATT à 2 ans confirment les très sérieux doutes de tolérance pour Avastin en ophtalmologie » (cote 1577). Le document conclut que « les patients traités par Avastin se voient injecter dans l'œil un produit n'ayant pas d'AMM pour cela alors même qu'il existe des alternatives validées par les autorités » (cote 1577).
Les « Counter Avastin Materials »
334. Dans un courriel du 17 février 2012, le siège de Novartis a adressé au groupe « Lucentis FIT » un document intitulé « Counter Avastin Materials » (cotes 2562 à 2572).
335. Ce document contient les arguments devant être utilisés par les salariés du groupe Novartis pour faire face à la publication des résultats à deux ans de CATT, ainsi que ceux des autres études en cours (IVAN et GEFAL) (« Counter-Avastin Materials. Preparing for 2yr [year] CATT & Similar studies (IVAN, GEFAL, etc.) », cote 2565).
336. Les actions devant être mises en œuvre, d’une part, par le siège suisse de Novartis, d’autre part, par les filiales régionales et nationales du groupe, sont exposées dans un tableau synthétique (cote 2567).
337. L’une des démarches envisagées est, à nouveau, d’insister sur les questions relatives au profil de sécurité d’Avastin. Le siège de Novartis devait donc fournir à ses filiales des informations sur l’étude Gower et les autres publications traitant de ce sujet, préparer la position du groupe sur les résultats de CATT à deux ans et fournir des éléments sur le changement de RCP d’Avastin (« Global (…) Gower publication, Avastin safety publications (…), Preparation and pre-position of CATT 2yrs [years] and IVAN results (…) Avastin label change (systemic ocular AEs [adverse effects] », cote 2567).
338. Les filiales régionales et nationales devaient, quant à elles, communiquer activement sur les données de sécurité d’Avastin et également sur les études CATT et IVAN. Elles devaient recruter localement des professionnels en faveur de l’argumentaire du groupe, soutenir la publication de données rétrospectives sur Avastin et contacter les KOL locaux pour générer des publications et une forte présence scientifique (« Regional & CPO (…) Active communication of Avastin safety data. Proactive positioning of CATT and IVAN results (…) Local engagement with KOLs to generate publications and scientific presence », cote 2567).
La mise à jour de l’« Elevator Speech » et le document « CATT : les messages clés »
339. Novartis a ensuite mis à jour son document intitulé « Elevator speech » qui contient la version synthétisée de l’argumentaire de l’entreprise sur la sécurité d’Avastin, pour y inclure les données issues des résultats à un an de CATT (cote 3102).
340. Le document indique notamment que « l'étude CATT, menée sur 1 208 patients indique une augmentation de 29 % du risque d'accidents systémiques sérieux nécessitant une hospitalisation avec Avastin comparé à Lucentis ». Il continue en affirmant que « ces écarts de tolérance sont probablement liés aux différences de structure moléculaire et à une exposition systémique plus importante sous Avastin ».
341. Un document additionnel, intitulé « CATT, les Messages clés », réalisé par Novartis, revient plus spécifiquement sur les résultats de l’étude CATT à un an (cote 3103).
342. Ce document indique que « Dans l'étude CATT, un plus grand nombre de décès et une augmentation statistiquement significative du risque d'accidents systémiques sérieux nécessitant une hospitalisation ont été observés avec Avastin comparé à Lucentis (29 % de risques supplémentaire avec Avastin pour les accidents systémiques sérieux. Ce signal en termes de tolérance est cohérent avec l'ensemble des résultats obtenus dans de précédentes études cliniques, notamment les larges analyses Medicare, indiquant que les risques de mortalité et d'accident vasculaire et cérébral sont plus élevés avec Avastin comparé à Lucentis. Les écarts de tolérance observés dans CATT entre Avastin et Lucentis sont probablement liés aux différences de structure moléculaire et d'exposition systémique consécutive aux injections intravitréennes ».
343. Dans ces deux documents, Novartis insiste donc sur le risque accru d’effets secondaires systémiques qui serait lié à l’utilisation d’Avastin, en liant l’existence de ces effets avec les différences de structure moléculaire entre Avastin et Lucentis.
Le document « Integrated Product Strategy (IPS) 2013 – Lucentis »
344. Dans un document, intitulé « Integrated Product Strategy (IPS) 2013 – Lucentis », daté de juin 2012, Novartis détaille les actions envisagées pour contrer les effets de la publication des études de comparaison en France (cotes 4483 à 4543).
345. Plus spécifiquement, Novartis prévoit de transmettre sa position aux autorités de santé (« Novartis position statement to Health Authorities », cote 4494), de mettre en œuvre un plan de communication vis-à-vis des KOL, des prescripteurs et du grand public (« Roll-out communication plan to the general public, KOL, prescriber and internal », cote 4494), de contrôler chacun des partenariats avec les associations de patients et de mettre en place des formations d’entraînement aux contacts avec les médias (« mapping of each partnerships w/patients/HCP associations + media training », cote 4494), l’objectif étant de discréditer la pratique de prescription « hors AMM », en préparation de l’impact anticipé de l’étude GEFAL (« Discard off-label use. Be ready to handle GEFAL impact if needed », cote 4494 ; voir également cote 4521).
La réunion « MAREG Franche-Comté » du 29 juin 2012
346. Une présentation établie en vue d’une réunion « MAREG(29) » pour la Franche-Comté, le 29 juin 2012, détaille les actions de Novartis dans cette région à cette époque (cotes 15696 à 15700).
347. Pour la franchise Ophtalmologie de Novartis, deux centres hospitaliers sont visés : le CHU de Besançon et le centre hospitalier de Vesoul.
348. Pour le CHU de Besançon, l’objectif affiché est de « transférer les unités d’Avastin utilisées dans ces indications en unités de Lucentis » (cote 15699). En effet, à cette époque, Avastin était largement utilisé dans les indications autres que la DMLA et Lucentis était en train d’obtenir deux nouvelles indications (OMD et OVR). Pour assurer ce transfert, il était notamment prévu que le MSL de Novartis présente les messages du groupe sur les résultats à deux ans de CATT (« Consolider l’opinion des médecins et du service pharmacie ; Lucentis différent d’Avastin sécurité d’emploi supérieure », cote 15700).
349. Pour le centre hospitalier de Vesoul, l’objectif était de « s’opposer au référencement d’AVASTIN en ophtalmo pour indications OMD et OVR » (cote 15699). Pour ce faire, les employés de Novartis devaient intervenir dans les COMEDIMs (Commissions du médicament et des dispositions médicaux) de l’établissement, et présenter les messages du groupe sur les résultats à deux ans de CATT (cote 15699).
La réunion « MAREG Bourgogne » du 3 juillet 2012
350. Une présentation établie en vue d’une réunion « MAREG » pour la Bourgogne, le 3 juillet 2012, détaille les actions de Novartis dans cette région à cette époque (cotes 15704 à 15707).
351. S’agissant en particulier du CHU de Dijon, l’objectif affiché est le suivant : « Augmenter la consommation de Lucentis dans les nouvelles indications maintenant remboursées. OMD très présent à l’hôpital. Transférer les unités d’Avastin utilisées dans ces indications en unités de Lucentis » (cote 15706).
352. Pour atteindre cet objectif, il est notamment prévu de développer des liens avec la pharmacie d’hôpital et de « Consolider l’opinion des médecins et du service pharmacie ; Lucentis diffèrent d’Avastin sécurité d’emploi supérieure. Contrat de bon usage du médicament. Vigilance PHMEV [prescriptions hospitalières des médicaments délivrés en ville] » (cote 15707).
353. Novartis prévoit en outre d’« avoir des alternatives privées / publiques locales au CHU de Dijon surpuissant pour Lucentis » (cote 15706).
La mise en œuvre du « Plan d’action Avastin » en septembre 2012
354. Un courrier électronique interne du 12 septembre 2012 revient sur la mise en œuvre du plan de Novartis vis-à-vis des KOL en région, prévu dans le document « Integrated Product Strategy (IPS) 2013 – Lucentis » susvisé (cotes 15017 à 15019).
355. À cette date, 22 KOL ont été listés dans les départements de l’Essone, des Hauts-de-Seine, de la Seine-St-Denis et du Val-de-Marne, Novartis prévoyant de les contacter afin de réaliser les actions suivantes : « Evaluation par les DH (ou MSL si opportunité) du niveau de connaissance qu’ont ces médecins des données de safety obtenues par l’Avastin lors de son utilisation dans le traitement de la DMLA => Deadline mi-octobre. Présentation par les MSL, sur sollicitation, des données disponibles, en particulier de safety => asap post sollicitation » (cotes 15017 et 150818).
356. Ainsi, la méthode de Novartis consistant à approcher les KOL sur le sujet de l’utilisation d’Avastin en ophtalmologie consistait à ne pas directement diffuser un discours remettant en cause la sécurité d’Avastin, mais plutôt à provoquer la discussion, en questionnant le professionnel de santé en cause sur son niveau de connaissance sur le sujet, pour ensuite lui fournir les arguments de l’entreprise lorsque l’intéressé en faisait la demande.
357. Un courriel interne à Novartis du 10 décembre 2012 montre que ce plan d’action était toujours en cours à cette date (« Chers tous, Je vous propose de faire un point sur l'état d'avancement du plan d'action Avastin (médecins ciblés par binôme DR/MSL) », cote 15021).
La réunion « LBT » (ou « Local Brand Team ») de décembre 2012
358. Un document intitulé « LBT [Local Brand Team] Lucentis – Main issues 2012-2017 », daté de décembre 2012, fournit un descriptif synthétique du plan d’action mis à jour par Novartis (cotes 3045 à 3054).
359. L’objectif de Novartis affiché dans ce document est de répondre à la menace que constitue Avastin, en particulier en créant, pour les organismes payeurs, un environnement dans lequel les produits « hors AMM » ne sont pas des comparateurs (« Objective & Deliverable (…) Manage Avastin threat and reposition it as an unjustified and illegal comparator for pricing negociation. An action plan involving has been set to create for payors an environment where unlicensed products are not comparators », traduction libre : « Objectifs et Résultats (…) Gérer la menace Avastin et le repositionner comme un comparateur illégal et injustifié pour la négociation sur le prix. Un plan d’action a été mis en place pour créer pour les organismes payeurs un environnement dans lequel les produits n’ayant pas d’AMM ne sont pas des comparateurs », cote 3048).
360. Les actions mises en œuvre incluaient des rendez-vous avec les autorités de santé sur la question du remboursement de Lucentis pour les indications en DME (« diabetic macular Edema ») et RVO (« Retinal vein occlusions »), un plan de communication dirigé vers les 83 principaux KOL, un plan de communication à long terme vis-à-vis des journalistes, en vue notamment d’anticiper la publication des résultats de GEFAL, la publication d’une déclaration par l’association Retina France et un plan de communication vis-à-vis des médecins et des pharmaciens (cote 3048).
Le compte rendu du directeur de la franchise Ophtalmologie de Novartis pour l’année 2012
361. Dans un compte rendu rédigé par le directeur de la franchise Ophtalmologie en vue de son évaluation individuelle pour l’année 2012, la gestion de la publication des résultats à deux ans de l’étude CATT est indiquée comme un objectif (« CATT 2 years results management », cote 45127).
362. Par ailleurs, parmi les résultats satisfaisants mentionnés dans ce document, figurent notamment le fait que 50 % des spécialistes de la rétine considèrent que la sécurité d’Avastin est inférieure à celle de Lucentis (« 50 % of RS [retina specalists] considering Avastin safety is inferior to Lucentis », cote 45128), la mise en place de partenariats avec les professionnels impliqués dans GEFAL (« partnership built with Gefal stakeholders », cote 45128) ou encore le fait qu’aucun leader d’opinion, ni aucune association de praticiens ou de patients, ne se soit prononcé publiquement en faveur d’Avastin, à l’exception de l’Hôtel-Dieu (« no public pro Avastin position by key physicians / physicians associations / patient associations except Hotel Dieu (post CATT 2 or post DGS letter) », cote 45128).
363. En revanche, le compte rendu note qu’il faut poursuivre la gestion de la communication sur la réforme législative des RTU, malgré un résultat décevant (« Management of the RTU law advocacy despite disappointing outcome », cote 45128).
f) Les éléments de communication au cours de l’année 2013
Le document « Lucentis 2013 draft IPS »
364. Un document, intitulé « Lucentis 2013 draft IPS », précise la stratégie générale du groupe Novartis concernant Lucentis (cotes 822 à 830).
365. Ce document indique que l’usage « hors AMM » d’Avastin demeure un défi important pour Novartis (« However, the challenge from unlicensed intravitreal Avastin remains and is likely to be exacerbated by further H2H data release in 2013/14 », traduction libre : « Toutefois, le défi de l’usage hors AMM d’Avastin en intravitréen demeure et sera sans doute exacerbé par les nouvelles diffusions de données issues d’études de comparaison en 2013/2014 », cote 827).
366. Le groupe Novartis prévoit ainsi de continuer à véhiculer des messages sur les risques de l’utilisation d’Avastin en termes de sécurité (« Medical affairs : Reinforce Lucentis as standard of care in wet AMD and continue driving messages of Avastin safety risks », cote 828), en revenant notamment sur les études de comparaison (« effective communication of emerging Avastin safety risks and methodological issues of H2H trials through internal (…) and external communication », cote 828).
367. De manière générale, il est demandé aux équipes des filiales nationales de continuer de diffuser le message du groupe sur les effets indésirables d’Avastin, en s’assurant de la plus grande couverture géographique possible, par le biais notamment du financement d’articles passant en revue les études de comparaison et une présence dans les congrès et symposiums (cote 828). À cet égard, il est indiqué que la France faisait à l’époque partie des pays les plus à risque du point de vue de Novartis (« Focus support on large high risk countries e.g. France, Germany », cote 828).
Le « Plan d’action GEFAL – PAG [Plan d’action Gefal]»
368. Un courriel interne à Novartis du 19 février 2013, et le compte rendu en pièce jointe, montrent qu’une réunion s’est tenue le 18 février 2013 au sein de Novartis, afin de préparer un « Plan d’action GEFAL – PAG [Plan d’action Gefal] » (cotes 15823 à 15826).
369. Ce document envisage un ensemble complet d’actions en lien avec la publication prochaine des résultats de l’étude GEFAL. Tout d’abord, Novartis prévoit une mise à jour, au regard des données récentes sur l’étude GEFAL et sur le projet de RTU, des documents contenant son argumentaire sur l’usage d’Avastin en ophtalmologie (« messages-clés » et « Q&A », cote 15825). Ensuite, les équipes de Novartis doivent être formées progressivement – du comité exécutif aux forces commerciales sur le terrain – afin de leur permettre de répondre aux questions qui seraient posées à la suite de la publication de GEFAL. Il est également prévu que les salariés de Novartis présents au congrès de l’ARVO de 2013 bénéficieront d’une formation spécifique (« Communication interne : ‘train to be ready for reactive answers’ (…) comité exécutif (…) Terrain : formation séminaire mars : design GEFAL + rappel CATT-IVAN 1 ; Messages clés avec résultats GEFAL et IVAN année 2 (…). Validation de ce qu’il est possible de diffuser », cote 15825).
370. S’agissant des actions de communication externes, le compte rendu précise que deux comités consultatifs seront organisés (l’un pendant le congrès de l’ARVO, l’autre juste après), que 17 KOL devant être contactés ont été identifiés, et que la Société Française d’Ophtalmologie, le Syndicat National des Ophtalmologistes de France ainsi que l’association de patients Retina France seront approchés. Concernant les journalistes, le compte rendu précise que les messages de Novartis doivent uniquement être diffusés de façon « réactive » (cote 15826).
371. Enfin, le compte rendu prévoit que le siège de Novartis sera tenu informé (« Communication au global Novartis : présentation ¾ slides avec vision globale (…) brief TVM sur les différentes actions (…) », cote 15826).
Le document « Messages clés Lucentis/Avastin » mis à jour en mars 2013
372. Dans un courriel interne du 11 mars 2013, Novartis revient sur le « Plan d’action publication GEFAL », visant à former les équipes commerciales en réaction à la publication prochaine des résultats de l’étude GEFAL (cotes 2980 à 2983).
373. En particulier, Novartis a mis à jour un document de « Messages clés Lucentis / Avastin » destiné à être distribué aux visiteurs médicaux, aux directeurs régionaux et aux employés du siège de l’entreprise (cote 2982). Ce document insiste à nouveau sur les risques liés au fractionnement et au reconditionnement nécessaires pour l’utilisation d’Avastin dans l’œil, sur les incertitudes qui demeureraient quant à la tolérance d’Avastin et enfin sur le fait que l’étude GEFAL n’a pas été dimensionnée pour lever définitivement les incertitudes sur la tolérance d’Avastin. Le document conclut en affirmant qu’il existe des alternatives validées par les autorités permettant de garantir la sécurité des patients (cote 2983).
374. En outre, la question de la lutte contre l’utilisation « hors AMM » d’Avastin fait partie du programme du séminaire « Ophta » organisé par Novartis au mois de mars 2013, au cours duquel sont prévus des ateliers « GEFAL » ou « Études hors AMM » (cote 4743).
La présentation du 10 avril 2013
375. Dans une présentation datée du 10 avril 2013, Novartis met à jour son plan d’action (cote 45146).
376. Dans ce document, Novartis envisage la mise en place d’un certain nombre d’actions afin de sécuriser les conditions de l’accès au marché de Lucentis (« Secure Lucentis market access conditions. Despite GEFAL and new RTU law, no compromise will be accepted regarding patients safety and Novartis rights », traduction libre : « En dépit de l’étude GEFAL et de la nouvelle loi RTU, aucun compromis ne sera accepté concernant la sécurité des patients et les droits de Novartis », cote 45146). Plus précisément, le plan d’action repose sur trois idées : (i) « ne faire aucun compromis sur la sécurité des patients » vis-à-vis des autorités de santé, ce qui implique notamment de diffuser à ces autorités et au LEEM les « messages clés du groupe » ou encore de préparer une stratégie contentieuse devant la Commission européenne et les juridictions nationales (« Be without compromise on Patient Safety and Novartis rights with Health Authorities (…) build & disseminate ‘one page key messages’ on Novartis position to HA [health authorities] and LEEM [Les Entreprises du médicament]. Legal complaint at EU (…) », cote 45146), (ii) « être centré sur la sécurité des patients » vis-à-vis des médecins et des associations de patients, notamment en formant les équipes commerciales sur le sujet et en diffusant le message du groupe auprès des KOL (« Be focused on patient safety with HCPs & Patient associations (…) Top KOL management by BU head (…) Detailed Q&A for FLM (…) », cote 45146) ; et (iii) y aller doucement avec les médias et le grand public, c'est-à-dire notamment éviter toute communication proactive, à l’exception du communiqué de presse de Novartis de réaction à la publication des résultats de l’étude GEFAL (« Be soft with Media & General public. NO proactive communication except post GEFAL Novartis statement », cote 45146).
La présentation « Lucentis H2H Action Plan » d’avril 2013
377. Une présentation, intitulée « Lucentis H2H Action Plan », datée d’avril 2013, confirme la stratégie poursuivie par le groupe Novartis concernant la concurrence d’Avastin en ophtalmologie (voir notamment cotes 14837 à 14848).
378. S’agissant plus particulièrement de la France, Novartis anticipe un risque de déplacement des patients des cabinets de ville vers les hôpitaux, où l’utilisation d’Avastin pourra avoir lieu dans le cadre d’une RTU (« Despite Avastin safety concerns, advocates exist in many markets. (…) France. Risk that treatment of patient will move from private practice (currently 85 % volume) to public hospitals where TRUs can be implemented », traduction libre : « En dépit des préoccupations concernant la sécurité d’Avastin, il y a des défenseurs sur plusieurs marchés (…) France. Risque que le traitement des patients soit assuré non plus dans les cabinets de ville (actuellement 85 % des volumes) mais dans les hôpitaux publics où la RTU pourra être mise en œuvre », cote 14833), susceptible d’avoir un impact sur les organismes payeurs (« Potential impact : National level pricing pressure. Risk of spill over to other markets », traduction libre: « Impact potentiel: pression sur les prix au niveau national. Risque d’extension à d’autres marchés », cote 14833).
379. Par ailleurs, cette présentation insiste sur les graves effets secondaires identifiés dans les études CATT et IVAN : « Pooled GATT & IVAN study showed significant increase in serious adverse events », cote 14841).
L’échange de courriels d’avril 2013 et le « plan d’action GEFAL auprès des ophta »
380. Un échange de courriels interne à Novartis en date du mois d’avril 2013 et la présentation annexée (cotes 15709 à 15711 ; cotes 15712 à 15754) montrent qu’une formation spécifique portant sur la réponse du groupe aux études de comparaison entre Lucentis et Avastin était prévue lors du prochain séminaire des directeurs régionaux.
381. S’agissant de la communication aux professionnels de santé, la présentation souligne l’importance de se concentrer sur les questions de tolérance (« Notre communication aux professionnels de santé. ‘Focus tolérance’. Même ligne de conduite que les précédentes années : 1. pas de communication proactive 2. Focus tolérance : -mêmes messages clés : ‘Différencier les 2 molécules’ –mise à jour en mai avec GEFAL » (cote 15718).
382. En outre, un « plan d’action GEFAL auprès des ophtas », avec un « focus tolérance » était prévu (cote 15722). Ce plan supposait essentiellement de former de manière intensive l’ensemble des équipes commerciales en contact avec les professionnels du secteur pour leur permettre de répondre à toutes les objections sur les résultats de GEFAL.
Le document « ARVO 2013 Global activity guide » de mai 2013
383. Un document, intitulé « ARVO 2013 Global activity guide », de mai 2013, présente l’ensemble du programme d’action prévu par Novartis au moment du congrès de l’ARVO de 2013, durant lequel les résultats de l’étude GEFAL devaient être présentés (cotes 15797 à 15821).
384. Ce programme prévoit notamment, le 7 mai, un « Joint Management Meeting » avec Genentech (cote 15807) et un « Scientific experts forum », durant lequel de nombreux médecins, dont 12 français, étaient invités (cotes 15807 et 15816).
385. Le 8 mai, un « Media webcast » est prévu. Novartis prévoit de publier un communiqué de presse sur les résultats à deux ans de l’étude IVAN (cote 15807). Le programme de ce « Media webcast », préparé par Novartis, indique que les objectifs de cette manifestation étaient de donner aux médias une mise à jour des dernières données concernant Lucentis, notamment par une présentation des études GEFAL et IVAN, pour continuer à différencier Lucentis et Avastin dans le traitement de la DMLA (« Provide media with an update of the latest Lucentis data ‘live’ from the congress : - Present and contextualise the GEFAL and IVAN head-to-head studies, to continue to clearly differentiate Lucentis vs Avastin in the treatment of wet AMD and proactively manage reporting around this topic - Communicate existing body of safety data from other key studies to strongly position Lucentis vs new-comer drug aflibercept », cote 15818).
386. Le 9 mai, Novartis prévoit de publier un communiqué de presse sur l’ensemble des résultats du congrès de l’ARVO. Un dîner avec les consultants invités dans le cadre du programme ACUITY est organisé, suivi le lendemain d’un comité consultatif ACUITY. Enfin, une campagne Twitter était prévue tout au long du congrès (cotes 15807 et 15817).
Le programme des équipes de Novartis pendant le congrès de l’ARVO
387. Un programme, intitulé « ARVO 2013 Program Schedule », établi par Novartis pour le congrès de l’ARVO de 2013 montre que des activités spécifiques ont été prévues par les équipes françaises du groupe (cotes 15830 à 15841).
388. À titre d’illustration, le 7 mai, Novartis organise un dîner à destination d’ophtalmologistes français invités, dont le thème est : « Différences entre les différents anti-VEGF avec l’intervention de KOL internationaux » (cote 15837). Le 8 mai, un comité consultatif (« Board ») DMLA est organisé, avec 11 KOL français (cote 15839).
Le compte rendu du comité consultatif national du 8 mai 2013
389. Le 8 mai 2013, le comité consultatif a eu lieu à Seattle, juste après la publication des résultats de GEFAL au congrès de l’ARVO (cotes 15001 à 15003).
390. Pendant cette réunion, le comité a analysé les résultats de GEFAL en détail. Le compte rendu indique sur ce point : « Safety : pas de différence significative entre les bras [c’est à dire l’ensemble de patients recevant le même traitement]- pas de nouveau signal d’alerte ; attention étude non dimensionnée pour faire ressortir les El [évenements indésirables] rares - Attention : 1 flacon /patient pour prévenir les risques d’endophtalmies » (cote 15001) et « Positionnement des membres sur cette étude : réussite de cette étude française - difficile de se prononcer sur la safety sur ces études non dimensionnées pour faire ressortir les El [effets indésirables] - une étude de safety serait nécessaire » (cote 15002).
391. Dans le cadre de cette discussion, les membres présentés ont également conseillé Novartis sur sa communication à suivre : « Conseils des membres : préparer les données de safety Avastin dans les études internationales - insister sur ‘1 flacon par patient’ dans GEFAL - rappeler l’importance des études montrant la baisse de cécité légale [c'est-à-dire le niveau de handicap visuel qui définit une personne aveugle] grâce aux anti VEGF dans la DMLA (type étude Danemark) » (cote 15003).
Le communiqué de presse de Novartis du 14 mai 2013
392. Le 14 mai 2013, soit une semaine après la publication des résultats de l’étude GEFAL, Novartis a publié un communiqué de presse destiné à la presse médicale et grand public (cotes 14997 et 14998, cote 2985).
393. Un échange de courriels internes (cotes 2988 à 2990) montre en particulier que les dirigeants de Novartis ont fait le choix délibéré de ne pas mentionner le résultat de l’étude dans leur communication et de ne mentionner que ses limites méthodologiques (« I have comments (…) - qualité méthodo (…) - rigueur de la recherche clinique franc[aise] – je ne répèterais pas le résultat : je dirais : l’étude conclut à la non infériorité sur un des critères d’évolution, sans pour autant aborder d’autres critères », cote 2988).
394. Le communiqué finalement rédigé reprend les critiques usuelles de Novartis sur le dimensionnement des études de comparaison au regard de la détection des effets secondaires et affirme que la méta-analyse des études CATT, IVAN, GEFAL et MANTA confirme « un risque plus important d'événements indésirables graves sous bevacizumab [Avastin] par rapport au ranibizumab [Lucentis] » (cote 2985). Novartis continue en indiquant que « seules des études sur de plus amples populations et à plus long terme pourraient écarter le doute qui pèse sur les effets secondaires du bevacizumab [Avastin] injecté dans l'œil - effets qui pourraient être majorés en pratique courante au regard de l'absence de présentation adaptée pour l'œil » (cote 2985).
395. Ce communiqué a été diffusé à l’ensemble de la presse médicale et pharmaceutique, tant généraliste que spécialisée, aux agences de presse grand public, en ce compris la presse santé et pour les seniors, comme en atteste le courriel interne de Novartis du 14 mai 2013 : « Le communiqué de presse Novartis vient d'être transmis à l'agence et sera diffusé dès ce soir à la presse médicale (MG, Pharmacien, Ophta, Onco, diabète, endocrino) ainsi qu'aux agences de presse grand public (Presse quotidienne nationale, hebdos. Presse santé. Presse senior. Radio et TV » (cote 2978). Il a été effectivement repris dans plusieurs publications destinées aux ophtalmologistes, comme en témoigne le courriel interne de Novartis du 13 juin 2013 : « Je vous prie de bien vouloir trouver ci-joint une nouvelle reprise du CP GEFAL parue dans le dernier Pratiques en Ophtalmologie » (cotes 15843 à 15844).
Le compte rendu du comité consultatif national du 22 mai 2013
396. Dans le cadre du compte rendu du comité consultatif national DMLA du 22 mai 2013, Novartis a inclus le communiqué de presse, cité ci-dessus (cotes 14997 et 14998).
397. Le compte rendu ajoute que « Il a aussi été rappelé que le décret d’application du RTU n’est toujours pas paru » (cote 14998).
Le document « Integrated Product Strategy (IPS) 2014 Lucentis / Ophtalmology » de juin 2013
398. Dans une présentation du 4 juin 2013, Novartis a mis à jour sa stratégie, après la publication des résultats de GEFAL (cotes 3005 à 3044). Novartis souligne notamment le risque que les autorités de santé accordent une RTU à Avastin, compte tenu de l’adoption en décembre 2012 de la « RTU économique » (« Secure Lucentis market access conditions & manage the risk of Avastin official endorsment in wAMD by HA (…) Economic RTU voted in December 12 (…) Legal complaint at EU (w/ global team) and national levels », traduction libre : « Sécuriser les conditions d’accès au marché de Lucentis et gérer le risque qu’Avastin soit officiellement régularisé dans le traitement de la DMLA par les autorités de santé (…) RTU économique votée en décembre 2012 (…) Contestation juridique aux niveaux européen (avec l’équipe du siège) et nationaux », cote 3041).
399. Dans ce document, Novartis relève le constat de non-infériorité et l’absence de différence en termes de sécurité d’Avastin par rapport à Lucentis (« Press : Communication on Avastin equivalence on efficacy and safety vs Lucentis », cote 3041).
400. Par ailleurs, Novartis envisage le cas où une RTU serait adoptée pour Avastin pour le traitement de la DMLA, soulignant que le risque principal de passage de l’utilisation de Lucentis à celle d’Avastin concernait le secteur hospitalier, qui représentait 25% des patients traités. En termes financiers, le risque de perte globale est évalué à 15 millions de dollars américains (« Impact'14 as of Q3'14 ->-32k units -$15m net sales vs Lucentis B'14 », cote 3041).
Les « Questions-Réponses Avastin / Lucentis » de juin 2013
401. Un document de « Questions / Réponses » sur le sujet Avastin / Lucentis, du mois de juin 2013, fournit le discours complet de Novartis, mis à jour après la publication des résultats de l’étude GEFAL (cotes 2510 à 2524).
402. Ce document précise qu’il est réservé à l’usage des directeurs régionaux de Novartis pour répondre aux questions éventuellement posées par des professionnels de santé (« Document interne exclusivement réservé à l’usage des directeurs régionaux Novartis SAS. Aucune diffusion ni utilisation extérieure », cote 2510).
403. L’un des ajouts récents (« New ») porte sur la réponse à apporter à l’affirmation suivante : « Aujourd’hui, avec l’étude française GEFAL en plus, il y a un niveau de preuve suffisant pour utiliser Avastin en toute tranquillité dans la DMLA » (cote 2515). La réponse préparée par Novartis reprend, pour l’essentiel, le contenu de son communiqué de presse du 14 mai 2013. Elle souligne en particulier qu’« Au même titre que CATT (1206 patients) et IVAN (610 patients), l’étude GEFAL n’apporte pas de preuve suffisante pour conclure sur le profil de sécurité d’Avastin dans la DMLA ». Elle revient sur le fait que « la méta-analyse regroupant notamment CATT, IVAN, GEFAL à 1 an, montre un risque d’effets systémiques graves augmenté de 34 % avec l’utilisation d’Avastin ». Elle conclut qu’« A ce jour, il est impossible de répondre à la question de la sécurité sanitaire des patients atteints de DMLA exsudative qui se verraient injecter Avastin dans l’œil », et en rappelant l’existence d’un médicament autorisé dans cette indication (« Il existe une alternative ayant une AMM dans la DMLA : Lucentis, dont le rapport bénéfice/risque est clairement établi », cote 2515).
404. Le document de « Questions / Réponses » contient en outre de nombreux arguments relatifs au risque en termes de responsabilité civile, pénale et disciplinaire pour un médecin qui prescrit Avastin, alors qu’il existe une alternative thérapeutique disposant d’une AMM (« le médecin engage sa responsabilité civile, pénale et disciplinaire s’il fait courir un risque injustifié au patient tandis qu’il existe une alternative thérapeutique disposant de l’AMM dans cette indication », cote 2512 ; voir également cote 2519).
405. Enfin, ce document revient, dans une partie intitulée « argumentaire détaillé », sur les projets de « RTU économique » en cours à l’époque, pour répondre à l’affirmation « Aujourd’hui, avec l’étude française GEFAL en plus, Avastin va obtenir une RTU dans la DMLA ». La réponse préparée par Novartis indique notamment que ce projet de « RTU économique » serait « Une mesure contradictoire avec les attentes de la société exprimées très fortement à l’occasion des derniers scandales sanitaires », soulignant le contexte des affaires du Mediator et de la pilule de 3ème génération, ainsi que le risque de mise en cause de la responsabilité des pouvoirs publics et des médecins (cote 2523).
Le compte rendu du directeur de la franchise Ophtalmologie de Novartis pour l’année 2013
406. Dans un compte rendu rédigé par le directeur de la franchise Ophtalmologie en vue de son évaluation individuelle pour l’année 2013 (cotes 45125 et 45126), la gestion des projets de « RTU économique » du Gouvernement et celle de la publication des résultats de GEFAL sont indiquées comme des objectifs principaux.
407. Parmi les résultats satisfaisants pour l’entreprise à ce sujet, est citée la première communication officielle de Novartis, rédigée en collaboration avec des KOL importants, mentionnant clairement les différences de sécurité entre Avastin et Lucentis.
408. De même, ce document mentionne, parmi les résultats satisfaisants, l’augmentation de la prise de conscience de ce sujet en France, visible dans les communications publiques sur GEFAL, les déclarations du président de l’ANSM au Sénat, le rapport de la CNAM à l’Assemblée nationale ou encore les articles dans les médias (« First official Novartis written communication clearly stating Lucentis vs Avastin safety difference, in cooperation with top KOLs », cote 45126). Son auteur estime que cette prise de conscience des autorités publiques sur les questions de sécurité pourrait expliquer que le décret d’application de la « RTU économique » n’ait pas encore été publié à l’époque et que les autorités de santé se réorientaient vers une négociation du prix de Lucentis plutôt qu’une généralisation de l’usage d’Avastin (« Awareness of these safety issues has really increased in France (…) which can explain that ERTU décret is still not published and that Health Authorities bodies are reorienting their actions towards a Lucentis price discussion more than an Avastin endorsment », cote 45126).
g) Les éléments de communication au cours de l’année 2014
La présentation « Plan d’action DMLA » de février 2014
409. Une présentation, intitulée « Plan d’action DMLA », discutée lors d’une réunion du 10 février 2014 interne à Novartis (cote 13797 à 13817), revient sur l’entrée sur le marché d’Eylea.
410. Cette présentation montre que l’arrivée de ce nouveau concurrent était devenue la cause principale d’inquiétude de l’entreprise.
Le document « Budget Book 2014 »
411. Dans un document intitulé « Budget Book 2014 - France », Novartis expose son analyse de l’évolution des ventes de Lucentis en France pour l’année 2014.
412. Parmi les « impératifs stratégiques », la question du risque d’un soutien accru des autorités de santé à l’usage d’Avastin et des résultats de GEFAL est mentionnée (cote 2834).
Les « questions/réponses » du mois de mars 2014
413. Un courrier électronique interne à Novartis du 13 mars 2014 indique qu’un document de « questions / réponses » sur le sujet Lucentis / Avastin a été transmis aux directeurs régionaux de l’entreprise (cotes 15679 à 15683 et 15768 et 15769). Ce document devait être utilisé uniquement pour répondre aux questions éventuellement posées par des professionnels de santé.
414. Ce document de « questions / réponses » (cotes 15770 à 15795) mettait à jour la communication du groupe à la suite de la publication de la condamnation de Roche et Novartis par l’AGCM. Il contenait en outre tous les éléments déjà mentionnés ci-dessus dans le document équivalent dans sa version de juin 2013, et reprend l’« argumentaire détaillé » mentionnant le changement de RCP d’Avastin (cote 15787).
Le compte rendu de visite du 10 février 2014
415. Enfin, un compte rendu de visite en date du 10 février 2014 montre que la question du prix de Lucentis et de sa comparaison avec celui d’Avastin continuait d’être soulevée par les ophtalmologistes injecteurs. Dans ce contexte, il apparaît que les délégués hospitaliers de Novartis continuaient de diffuser les messages clés du groupe (cotes 15756 à 15757).
h) Conclusion
416. Il ressort des éléments qui précèdent que Novartis a mis en place une vaste campagne de communication et d’influence dont l’objectif était de lutter vigoureusement contre l’usage d’Avastin en ophtalmologie, en insistant de façon systématique sur les risques liés à l’utilisation « hors AMM » d’Avastin dans le traitement de la DMLA, en comparaison avec la sécurité garantie de Lucentis. Le laboratoire a, dans ce cadre, régulièrement mis à jour ses supports de communication au regard des évolutions scientifiques et réglementaires sur le sujet, et a utilisé l’ensemble des canaux à sa disposition (KOL, équipes commerciales sur le terrain, presse).
417. Ce discours a porté plus particulièrement sur les risques liés à l’utilisation d’Avastin « hors AMM » en ophtalmologie, alors qu’il existait une alternative thérapeutique disposant d’une AMM.
2. LES PRATIQUES MISES EN ŒUVRE PAR ROCHE ET NOVARTIS, AVEC L’AIDE DE GENENTECH, AUPRES DES POUVOIRS PUBLICS
418. Les laboratoires Novartis et Roche, en collaboration avec Genentech (a), ont développé un discours concernant l’utilisation d’Avastin en ophtalmologie, auprès des autorités publiques (b). Ce discours a évolué au cours du temps, en fonction de l’évolution du contexte scientifique et réglementaire, exposé ci-dessus (cf. paragraphes
92 à 171 ci-dessus).
a) Les relations entre Genentech, Roche et Novartis sur la question de l’usage d’Avastin en ophtalmologie
419. Comme exposé aux paragraphes 198 à 221 de la présente décision, la structure contractuelle liant Genentech avec ses deux preneurs de licence – Roche pour la spécialité Avastin et Novartis pour la spécialité Lucentis – confère au laboratoire américain une transparence totale de l’information et une certaine forme de contrôle sur les actions entreprises par ses licenciés pour la commercialisation d’Avastin et de Lucentis dans le monde. En outre, Genentech conserve une forte influence sur le plan de développement des indications de chaque spécialité.
420. Au-delà des relations entre ces trois entités découlant des contrats de licence, un certain nombre de pièces du dossier fournissent des informations sur la façon dont les relations entre Genentech et ses co-contractants étaient effectivement gérées et sur l’organisation des compétences et les modalités d’adoption de décision concernant les spécialités, entre les différentes entités impliquées.
Les relations entre Roche et Genentech
421. Il découle de plusieurs échanges entre les équipes de Roche et de Genentech que les décisions relatives à la stratégie à adopter s’agissant de l’utilisation d’Avastin en ophtalmologie étaient prises au sein de Genentech.
422. Un message du 28 avril 2011 adressé par M. M…, dirigeant de Genentech, sur une application web interne au groupe Roche, confirme cette situation. Dans ce message, il commente les résultats à un an de l’étude CATT et indique, à l’attention de l’ensemble du groupe Roche, que, selon lui, ces résultats confirment le choix de Genentech de se concentrer sur Lucentis dans le domaine ophtalmologique (« Based on our preliminary evaluation of the data, the outcome confirms our belief that Lucentis is the most appropriate treatment option », cote 6930).
423. De même, une note manuscrite du 10 mai 2011 de Mme C…, pharmacien responsable de Roche, comporte les indications suivantes relatives à Avastin : « DMLA en attente du position paper [document stratégique] de GNE » (cote 5349).
424. Ou encore, il ressort de plusieurs échanges entre les dirigeants respectifs de Roche en France, du siège de Roche et de Genentech que la décision de ne pas étendre l’AMM d’Avastin à couvrir la DMLA est adoptée par Genentech. En effet, dans un courriel du 30 juillet 2012, M. M…, le dirigeant de Genentech, souligne « Could you let Q… know that there is no way we will expand the Avastin label (…) to include AMD. As a manufacturer we would refuse it as we cannot accept the responsibility that goes with it » (traduction libre : « Peux-tu dire à Q… qu’en aucune manière nous n’étendrons l’indication d’Avastin pour le traitement de la DMLA. En tant que fabricant nous refuserions dès lors que nous ne pouvons pas accepter la responsabilité qui en découlerait » cote 7148). Celui-ci a donc enjoint ensuite aux équipes de Roche de cesser de se préoccuper de la question d’une éventuelle AMM pour la DMLA et de plutôt communiquer auprès des pouvoirs publics français sur les défauts de sécurité d’Avastin quand il est utilisé dans l’œil (« So French team do not need to spend time on checking how the indication can be granted and the label modified, they should rather go back to the government and explain to the above, in particular the lack of safety of Avastin when used in the eye », cote 7148 ; également cote 14689).
425. De même, en réponse à une interrogation d’un employé de Roche, D…, concernant une éventuelle extension de l’indication d’Avastin en ophtalmologie (« should we push for an extension of Avastin indication in ophtalmology for AMD or not in EU ! », cotes 14726 et 14727), Mme E…, employée de Genentech, indique dans un courriel du 3 septembre 2012 : « interesting but we’ve consistently said that we have no intention of developing Avastin for AMD or any other eye condition » (traduction libre : « intéressant mais nous avons dit de manière constante que nous n’avions pas l’intention de développer Avastin pour le traitement de la DMLA ou de toute autre indication ophtalmologique », cote 14726).
426. Par ailleurs, des notes manuscrites et des courriels saisis au sein des locaux de Roche montrent que les courriers de réponse de la société aux demandes de l’ANSM sur l’utilisation d’Avastin étaient revus par l’équipe internationale de Roche, qui fournissait les éléments scientifiques à insérer dans l’argumentaire, avec l’aide de Genentech (« Avastin DMLA – 6/09/2012. Teleconf avec le global regl. Pour mise au point des réponses à apporter. L’équipe internationale veut revoir le courrier », cote 5382 ; voir également cotes 14698 à 14701 : « vous trouverez ci-joint un projet de lettre au Pr F… sur le sujet des différences moléculaires et non cliniques d’Avastin et Lucentis (…) + tableau fourni par Genentech en annexe »).
427. Enfin, lorsque les équipes de Roche se sont rendues à un rendez-vous à l’ANSM au mois de novembre 2012, elles étaient accompagnées d’un employé de Genentech (cotes 14695 et 14696). Un courriel interne montre que cet employé fournissait les données relatives à Avastin, alimentant les discussions entre Roche et l’ANSM (« Merci G… pour l’invitation mais plutôt pour le 30/11 (même jour que réunion avec F…). J’espère que les données Avastin ont été utiles pour votre discussion », cote 14703).
Les relations entre Novartis et Genentech
428. Il découle de plusieurs courriels, saisis dans les locaux de Novartis, que des informations sur l’utilisation d’Avastin en ophtalmologie pouvaient être transmises directement par Roche à Novartis ou par l’intermédiaire de Genentech.
429. À titre d’illustration, dans un échange de courriels du mois d’août 2012, en réponse à une demande de Genentech, Roche a transmis à ce dernier des éléments portant sur le changement de RCP d’Avastin, informations qui ont été retransmises le même jour par Genentech à Novartis, alors qu’elles n’étaient pas encore rendues publiques (« As requested by Ralph [Genentech] please find hereattached the smPC/PL for II/4444 as agreed with the CHMP. The revised product information will be publicly available on the EMA website after the Commission Decision (September/October) », traduction libre : « Comme demandé par Ralph [Genentech], veuillez trouver ci-joint le smPC/PL for II/4444 tel que décidé avec le CHMP. Le RCP modifié sera rendu public sur le site de l’EMA après la décision de la Commission (septembre/octobre) », cote 15760 ; « Please see attached the forthcoming safety update to the European Avastin label which is expected to be finalised in Sept/Oct. Importantly there is mention of both local and systemic side effects associated with unlicensed intravitreal use. We will update the relevant CPO material for rapid distribution once this is finalised and public », (traduction libre : « Veuillez trouver ci-joint la mise à jour à venir du RCP d’Avastin qui devrait être finalisée en septembre/octobre. Il est important de relever la mention de l’existence d’effets indésirables locaux et systémiques associés à l’utilisation ‘hors AMM’ [d’Avastin]. Nous mettrons à jour les documents pour une distribution rapide quand cela sera finalisé et rendu public », cote 15759).
430. De même, au moment où les résultats de l’étude GEFAL ont été rendus publics, en mai 2013, un message de Mme H…, responsable du produit Lucentis chez Genentech, à plusieurs destinataires au sein du groupe Roche en date du 8 mai 2013, indique : « We're meeting with NVS at 6pm to discuss and align as much as possible on media strategy and messaging.
We anticipate they will be inclined to take an aggressive approach in the media based on potential impact of the GEFAL data on access to Lucentis in France. We anticipate possible media reaction and government pressure on Roche regarding Avastin in France as well » (traduction libre : « Nous rencontrons NVS [Novartis] à 18h pour discuter et nous aligner le plus possible sur notre stratégie médias et sur nos messages. Nous anticipons qu’ils auront tendance à adopter une approche agressive dans les médias, autour de l’impact potentiel des données GEFAL sur l’accès de Lucentis en France. Nous anticipons une possible réaction des médias et une pression du gouvernement sur Roche au sujet d’Avastin en France », cotes 14734 à 14736).
431. Ce message precise que si l’équipe procède à des modifications mineures pour mettre à jour les messages clés, elle a estimé qu’aucun changement de la stratégie et du message d’ensemble n’était nécessaire (« Team is currently making minor revisions to update some details in the key messages but has determined no change in our strategy and overall message is required », cote 14736).
b) Les échanges de Roche et Novartis avec les autorités publiques
Les échanges entre Roche et l’AFSSAPS sur la possibilité d’une étude de stabilité au cours de l’année 2008
Le contexte des premiers échanges entre Roche et l’AFSSAPS : la mise en place de l’étude GEFAL
432. Comme indiqué par un communiqué de presse de l’AFSSAPS, des discussions portant sur l’usage d’Avastin en ophtalmologie en France existent au moins depuis l’été 2006.
433. À cette époque, le comité de qualification des protocoles thérapeutiques de l’AFSSAPS avait été saisi de la question de l’utilisation d’Avastin dans le traitement de la DMLA. Ce comité avait alors conclu que la mise en place d’un protocole thérapeutique temporaire (PTT) pour Avastin pour la DMLA n'était pas justifiée devant l'insuffisance de données disponibles et l'absence de forme pharmaceutique adaptée à un usage intravitréen. Le comité plaidait donc pour la mise en place d’un essai comparatif de non infériorité versus Lucentis.
434. Au mois de février 2007, l’AFSSAPS a donc « engagé une réflexion sur l'utilisation de l'anticancéreux Avastin (bévacizumab, Roche) dans le traitement de la dégénérescence maculaire liée à l'âge (DMLA) ». L’AFSSAPS ajoutait : « on voudrait pouvoir disposer d'une étude comparative entre Lucentis et Avastin. Nous réfléchissons aux moyens nous permettant d'en disposer » (communiqué de presse de l’AFSSAPS, cotes 2019 à 2020).
435. Ainsi, à partir du moment où une réflexion sur l’usage d’Avastin dans la DMLA a été mise en place en France, et notamment au sein de l’AFSSAPS, celle-ci a contacté Roche afin d’obtenir un certain nombre d’informations complémentaires.
Les demandes d’informations de l’AFSSAPS et les réponses de Roche
♦ Le courrier de l’AFSSAPS du 27 février 2008
436. Le 27 février 2008, l’AFSSAPS a contacté par courrier Roche afin d’obtenir des échantillons d’Avastin, ainsi qu’un certain nombre d’informations et d’éléments chimiques, en vue de conduire une étude de stabilité(30) d’Avastin (cote 16398).
437. Le courrier précise que l’étude de stabilité en question avait pour but de sécuriser l’étude GEFAL mise en place : « Cette étude a été décidée par l’AFSSAPS compte tenu de la mise en place d’un essai clinique ‘’GEFAL’’ (promoteur : Hospices civils de Lyon) qui compare l’utilisation d’Avastin en ophtalmologie (en seringue pré-remplie) à un autre médicament dans le traitement de la DMLA. Cette étude de stabilité vise à vérifier que la mise en seringue du produit susceptible de provoquer un stress mécanique avec apparition d’agrégats plus ou moins réversibles, ne modifie pas sa stabilité » (cote 16398).
♦ La réponse de Roche du 7 avril 2008
438. Par un courrier du 7 avril 2008, Roche a refusé de donner suite à la demande de l’AFSSAPS (cotes 16400 à 16401).
439. Dans son courrier, Roche invoque la « situation délicate » à laquelle le laboratoire est confronté, soulignant plus spécifiquement les inquiétudes du comité des médicaments à usage humain (ci-après, « CHMP ») concernant l’usage « hors AMM » d’Avastin, le fait que Roche n’aurait pas la possibilité de réaliser des études cliniques pour le traitement de la DMLA, en raison du fait que le produit n’aurait été conçu que pour traiter le cancer, ou encore l’existence de problèmes de contamination liés au reconditionnement d’Avastin sous forme de seringues.
440. En conclusion, Roche affirme que son refus repose principalement sur son souhait de ne pas être considéré comme ayant approuvé l’usage « hors AMM » d’Avastin : « Vous comprendrez que nous soyons extrêmement réservés quant à la poursuite d’essais portant sur Avastin pour une indication DMLA. Comme vous le savez, notre inquiétude principale est qu’en répondant favorablement à votre demande de fourniture nous puissions être considérés comme approuvant cet usage hors AMM », cote 16401).
♦ Le courriel de l’AFSSAPS du 10 septembre 2008
441. Le 10 septembre 2008, le Dr I…, Chef du service de l’évaluation, de la surveillance du risque et de l’information médicale de l’AFSSAPS, a demandé de nouveau à Roche les échantillons nécessaires à la réalisation des études de stabilité de la solution Avastin reconditionnée en seringue.
442. Ce courriel insiste sur l’importance de communiquer ces informations, compte tenu de l’usage « hors AMM » d’Avastin constaté en pratique : « (…) tu dois savoir que nous avons en cours une réflexion sur l'utilisation hors AMM en ophtalmologie. Nous souhaitons réaliser à la DLC, plusieurs essais de stabilité de la solution reconditionnée en seringue. Nous avons besoin d'échantillons pour ce contrôle ... ainsi que les éléments indispensables pour les réaliser, en particulier les réactifs spécifiques, les références standard ... J'ai cru comprendre qu'une demande avait déjà été faite par la DLC. Pourrais-tu m'aider et me transmettre un point de contact car je pense que ces contrôles sont absolument nécessaires, en raison de l’utilisation importante hors AMM du produit » (cote 16403).
♦ Le nouveau refus de Roche le 28 novembre 2008
443. Par courrier du 28 novembre 2008, Roche a, de nouveau, refusé de communiquer les informations et échantillons demandés par l’AFSSAPS, en réitérant sa position exposée dans son courrier du mois d’avril 2008 (« Comme suite à notre courrier, (…), nous réitérons notre position et souhaitons vous rappeler la situation délicate à laquelle nous sommes confrontés : (…). Vous comprendrez donc que nous restions extrêmement réservés quant à la poursuite de tests portant sur Avastin pour une utilisation hors AMM en ophtalmologie. Notre inquiétude principale est qu'en répondant favorablement à votre demande, nous puissions être considérés comme approuvant cet usage hors AMM », cotes 16405 à 16406).
♦ La transmission des échantillons à l’AFSSAPS
444. Ce n’est que le 22 juin 2009, soit un an et quatre mois après la première demande de l’AFSSAPS, que le siège bâlois du groupe Roche a finalement transmis à l’AFSSAPS les éléments demandés (cotes 16408 et 16409).
La communication de Roche et Novartis après la publication des premiers résultats de CATT au cours de l’année 2011
445. Pendant l’année 2011, Roche et Novartis ont développé une communication réagissant à l’annonce des premiers résultats de l’étude CATT, insistant plus particulièrement sur les risques d’effets secondaires graves pour les patients traités par Avastin en injection intravitréenne.
Les « messages clés » de Roche
446. Un document de Roche, daté du 6 mai 2011, expose les « messages clés » en réaction à la publication de l’étude CATT (cotes 6932 et 6933), qui ont été mis à jour le 9 mai 2011 (cotes 6938 à 6942).
447. Dans ce document, Roche résume les résultats de l’étude CATT. S’agissant plus spécifiquement des critères de sécurité, il indique : « le risque d'effets secondaires systémiques serait moins important avec injection intra vitréenne de Lucentis comparé à Avastin » (cote 6932).
448. Par ailleurs, Roche expose sa position sur l’utilisation « hors AMM » d’Avastin en ophtalmologie. Il souligne en particulier que « Avastin et Lucentis sont deux médicaments différents » et, à deux reprises, que « Lucentis est le traitement le plus approprié ». Le laboratoire rappelle en outre la position de prudence recommandée par l’AFSSAPS dans son point d’information de septembre 2009 (cote 6933).
Le compte rendu de la réunion « Responsabilité pharmaceutique » du 9 mai 2011
449. Un compte rendu d’une réunion intitulée « Responsabilité Pharmaceutique » tenue au sein de Roche, le 9 mai 2011, indique, au sujet de l’utilisation d’Avastin dans le traitement de la DMLA : « Suite à la publication vendredi 6 mai d’un communiqué de presse sur Avastin dans la DMLA, des téléconférences avec la maison-mère ont été organisées afin de voir quelle position prennent Novartis et Roche » (cote 5334).
450. Interrogés en audition sur ce point, les représentants de Roche ont indiqué : « Pour moi le communiqué n'est pas un communiqué de Roche, mais plutôt une communication extérieure.
Dès lors que cela concernait l'AMM, il convenait d'en informer la maison mère, titulaire de l'AMM » (cote 16740).
Les échanges de Roche avec l’ANSM au sujet du développement d’Avastin pour le traitement de la DMLA
♦ Le courrier de l’AFSSAPS du 16 mai 2011
451. Le 16 mai 2011, l’AFSSAPS a adressé un courrier à Roche, en lui demandant quelles étaient ses intentions concernant l’usage d’Avastin pour le traitement de la DMLA (« Je souhaiterais connaître les intentions de votre société concernant l’usage de votre spécialité Avastin (bévacizumab) dans le traitement de la DMLA », cote 6876).
452. Dans ce courrier, l’AFSSAPS indique notamment que compte tenu de l’usage « hors AMM » d’Avastin en France, l’institution envisageait la mise en place d’un PTT pour Avastin si Roche décidait de déposer une demande d’AMM pour une indication dans le traitement de la DMLA (« Compte tenu de l’utilisation en dehors de l’autorisation de mise sur le marché (AMM) d’Avastin dans la DMLA en France, nous serions prêts à examiner la mise en place d’un protocole thérapeutique temporaire (PTT), sous réserve que les laboratoires Roche s’engagent à déposer, dans un avenir proche, une demande d’AMM dans cette indication avec une présentation adaptée à cet usage », cote 6876).
♦ La réponse de Roche du 26 mai 2011
453. Dans un courrier de réponse du 26 mai 2011, Roche réitère sa position sur le sujet (cotes 16424 à 16426).
454. Roche indique ainsi que « un programme de développement d’Avastin dans la DMLA ou autre maladie ophtalmique ne semble pas approprié » (cote 16424). Au soutien de son argumentaire, elle renvoie aux résultats des études CATT et Gower, plus particulièrement s’agissant des effets indésirables relevés pour Avastin.
455. Roche en conclut que le risque d’événements indésirables graves est potentiellement plus élevé pour Avastin (« Ainsi, les résultats de ces études montrent des différences potentiellement importantes entre les profils de tolérance d’Avastin et de Lucentis dans le traitement de la DMLA. Le risque d’événements indésirables graves est potentiellement plus élevé lors d’une utilisation hors AMM intra-vitréenne d’Avastin », cote 16425) en comparaison avec Lucentis, dont le profil de tolérance est bien établi (« Lucentis a été rigoureusement étudié dans 18 essais cliniques ayant inclus plus de 7 100 patients. Son profil de tolérance est bien établi, il a été autorisé dans le traitement de la DMLA par diverses autorités de santé, dont l’EMA », cote 16425).
456. En conclusion, Roche indique être « convaincu que Lucentis est le traitement le plus approprié » et que le laboratoire n’envisage pas de développer Avastin en ophtalmologie (cote 16426).
457. À ce sujet, un échange de courriers électroniques en date des 17 et 18 mai 2011 montre que le projet de courrier de réponse de Roche à l’ANSM, en version anglaise, avait été transmis, pour commentaires, au Docteur B…, alors vice-président de la commission d’AMM de l’AFSSAPS (cotes 6943 à 6945).
458. Or, celui-ci a critiqué l’analyse de Roche dans ce projet. Concernant notamment les différences en termes d’effets indésirables systémiques, le Docteur B… indique « en fait pas de différence sur les événements vasculaires ou infectieux, seule différence sur les ‘autres événements systémiques et en particulier gastro’ qui mélangent tout (hémorragies digestives avec nausées…) qui ne sont pas tous sévères et qui (pour des comparaisons multiples) ne conduit qu’à un p = 0,04. Pour des comparaisons multiples on est en droit de diviser la valeur du p par le nombre de comparaisons et alors ça n’est plus statistiquement significatif !!! ». Plus loin, le Docteur B… ajoute : « EMA OUI MAIS POUR UNE COMEDIMS ET DES ACHETEURS HOSPITALIERS CES EVENTUELLES DIFFERENCES (J'AI BIEN DIT "EVENTUELLES") PEUVENT NE PAS SUFFIRE POUR JUSTIFIER UNE TELLE DIFFERENCE DE PRIX » (cote 6945).
Le courrier de Novartis du 9 mai 2011 adressé aux acteurs institutionnels du secteur de la santé
♦ Le courrier du 9 mai 2011
459. Le 9 mai 2011, Novartis a envoyé un courrier à un grand nombre d’acteurs institutionnels du secteur de la santé, parmi lesquels figurent les principales autorités de santé (CEPS, AFSSAPS, HAS, ministère de la santé), la présidence de la République, les services du Premier ministre, le ministère de l’économie, les associations de patients, l’Ordre des médecins, l’Ordre des pharmaciens ou encore des associations d’ophtalmologistes (cotes 3082 et 3083).
460. L’objectif de ce courrier était de répondre à un article paru le 6 mai 2011 dans le journal Le Figaro, dont le titre était le suivant : « Cécité : deux molécules aussi efficaces, l'une à 30 euros, l'autre à 1200 » (cotes 3080 à 3083).
461. Dans ce courrier, Novartis indique que l’étude CATT, dont les résultats à un an viennent d’être publiés, a montré un risque d’accident systémique sévère nécessitant une hospitalisation augmenté de 29 % avec Avastin, par rapport à Lucentis. Le laboratoire ajoute que « la tolérance d’Avastin apparaît inférieure à celle de Lucentis de façon statistiquement significative, bien que les auteurs notent que la puissance statistique est insuffisante pour détecter des effets secondaires sérieux » (cote 3080).
462. Novartis cite ensuite les études rétrospectives Curtis et Gower, en soulignant les éléments relevés pour Avastin : (i) constat d’une augmentation du risque de mortalité et d’accident vasculaire cérébral hémorragique, dans l’étude Curtis ; (ii) constat d’une augmentation du risque de mortalité, d’accident vasculaire cérébral hémorragique et d’inflammation intraoculaire, dans l’étude Gower.
463. Novartis affirme enfin que « les différents signaux de tolérance liés à l’utilisation hors AMM d’Avastin en injection intra-vitréenne aujourd’hui identifiés dans les trois études précitées sont sérieux et ne doivent pas être négligés » (cote 3081).
464. En conclusion, Novartis invite les autorités publiques à « prendre position sur les incitations médiatiques à l’utilisation hors AMM d’Avastin » en ophtalmologie, le laboratoire pharmaceutique estimant que « le message porté par les médias est une incitation à l’utilisation hors AMM d’Avastin en ophtalmologie alors qu’il existe une alternative thérapeutique validée. Il pourrait entraîner des risques pour les patients et avoir des conséquences importantes en termes de sécurité sanitaire » (cote 3081).
♦ L’impact du courrier de Novartis dans le secteur de la santé
465. Le courrier de Novartis a eu un impact significatif dans le secteur de la santé en France.
466. Ainsi, le 11 mai 2011 Novartis se félicitait de ce qu’une dépêche APM (site d’information spécialisé dans le domaine de la santé) reprenait la quasi-totalité de ses arguments sur la différence entre Lucentis et Avastin (cote 3072).
467. De même, le 20 mai 2011, Novartis a reçu un courrier de réponse de l’Ordre des médecins lui indiquant : « Nous avons pris bonne note que vous avez saisi les autorités sanitaires chargées de la vigilance en France. Je vous remercie de bien vouloir nous communiquer pour information les réponses qui vous seront adressées » (cote 3085).
468. Le 24 mai 2011, le chef de la Direction de l’évaluation des médicaments et des produits biologiques (« DEMEB ») de l’AFSSAPS a répondu à la lettre de Novartis : « C'est avec la meilleure attention que j'ai pris connaissance de la problématique évoquée, en particulier du risque de prescription hors AMM de la spécialité Avastin, lors même que votre spécialité Lucentis bénéficie d'une autorisation de mise sur le marché dans le traitement de la DMLA cardiovasculaire. Je comprends parfaitement votre position sur le sujet et de ce dossier est en cours d’étude actuellement à la DEMEB. Nous ne manquerons pas de revenir vers vous dans les meilleurs délais » (cote 3064).
469. Le 7 juillet 2011, Monsieur F…, directeur général de l’AFSSAPS, a répondu en ces termes : « Etant particulièrement soucieux de la sécurité des patients, en l'absence d'évolution du dossier d'enregistrement d'Avastin, l'Agence entend rester sur la position de prudence prise en 2009, position qui rappelle notamment les incertitudes de cet usage au regard de la sécurité des patients du fait d'une forme pharmaceutique inadaptée à cet usage et de données de sécurité limitées » (cote 3063). Le courrier ajoutait cependant « Il apparaît que le prix de Lucentis génère une utilisation hors-AMM d'Avastin en ophtalmologie, situation qui pose un problème de santé publique auquel il conviendrait de trouver rapidement une solution » (cote 3063).
La communication de Genentech, Roche et Novartis au moment de l’annonce des projets de RTU par le gouvernement au cours de l’année 2012
470. Pendant l’année 2012, à partir du moment où les pouvoirs publics ont annoncé étudier la possibilité d’une « RTU économique » pour Avastin dans le traitement de la DMLA, Roche et Novartis ont multiplié les initiatives et les contacts avec les représentants du gouvernement et des autorités de santé, diffusant ainsi leur discours sur les risques associés à l’utilisation d’Avastin en ophtalmologie.
Les échanges entre Roche, Genentech et Novartis
♦ Les courriels du 30 juin 2012
471. Le 30 juin 2012, les équipes de Roche ont informé le siège du groupe du projet des pouvoirs publics d’étudier une RTU pour Avastin dans le traitement de la DMLA (cotes 14706 à 14707).
472. En réponse, un employé de Genentech, dont la fonction est « LifeCycle Leader Avastin », incluant le suivi de l’usage d’Avastin en ophtalmologie (« I am (…) LifeCycle Leader Avastin [responsable du cycle de vie Avastin] LC, BC and FA (including ophtalmic use) », cote 14706) a précisé que la position de Genentech était inchangée depuis l’année dernière, en renvoyant à des documents portant sur l’usage « hors AMM » d’Avastin dans le traitement de la DMLA et à l’envoi par Roche d’une lettre aux autorités de santé françaises (« Please find attached several documents outlining our position on the off-label use of Avastin in wAMD. Our position has not changed since last year (when the letter was sent by Roche France to the French HA [Health Authorities]) », traduction libre : « Veuillez trouver ci-joint plusieurs documents décrivant notre position concernant l’usage ‘hors AMM’ d’Avastin dans le traitement de la DMLA. Notre position n’a pas changé depuis l’année dernière (quand la lettre a été envoyée par Roche France aux autorités de santé françaises) », cote 14706).
♦ Les courriels du 2 juillet 2012
473. Faisant suite à cet échange, Roche a organisé, le 2 juillet 2012, une téléconférence « avec les équipes médicales de GNE [Genentech] afin que nous soyons très au clair sur les data disponibles » (cote 14712).
474. Le même jour, un courriel interne à Genentech, envoyé par le « LifeCycle Leader Avastin » mentionne cette conférence téléphonique avec Roche (« Today we had a TC [telephonic conference] with Roche France regarding Avastin and its off-label use in wAMD », traduction libre : « Aujourd’hui, nous avons eu une conférence téléphonique avec Roche France concernant Avastin et son utilisation ‘hors AMM’ en DMLA », cote 15766).
475. Ce courriel interne fait état des prochaines étapes envisagées par Roche et Genentech (« Next steps for Roche/GNE », cote 15766), distinguant une « équipe Avastin » (« Avastin team ») et une « équipe Lucentis » (« Lucentis team »). Il est ainsi prévu que l’équipe Lucentis informe Novartis de la situation et que l’équipe Avastin commence à évaluer quelles informations sur l’usage d’Avastin dans la DMLA devaient être transmises aux autorités françaises. L’équipe Lucentis devait également fournir des conseils sur le contenu des informations de sécurité et de tolérance devant être transmises aux autorités, s’agissant tant d’Avastin que de Lucentis.
♦ La transmission de ces échanges à Novartis
476. Le contenu de ces discussions internes à Genentech a ensuite été transféré aux membres du « Joint Management Committee » (« JMC ») pour Lucentis, incluant les représentants de Novartis. Le siège de Novartis a ensuite transmis ces éléments à Novartis (« Je viens d’être contacté par le Global qui a été informé par Roche/Genentech sur le process RTU Avastin », cote 15764).
477. Cette chaîne structurée de communication a été complétée par un certain nombre de contacts directs entre Roche et Novartis, qui s’informaient essentiellement de leurs démarches vis-à-vis des pouvoirs publics, en particulier au sein du LEEM.
478. À titre d’illustration, dans un courriel interne d’octobre 2012, une employée de Roche indique : « Suis au LEEM (…) Suis assise à côté de Novartis qui fera des recours au niveau européen si cela ressort dans un autre texte » (cote 14719). De même, dans un courriel interne d’août 2012, dont l’objet est « Document de référence Avastin – Lucentis », un employé de Roche indique « J… vient juste de me dire que tu as échangé avec Z… [PDG de Novartis] ce matin, donc je pense que vous êtes alignés » (cote 14732 ; voir également cotes 14738, 7135, 7142 et 44698).
La communication de Roche et Novartis auprès des acteurs institutionnels
♦ La déclaration du président de la HAS du 29 juin 2012
479. Dans une déclaration à l’agence de presse médicale (« APM ») le 29 juin 2012, M. X…, le président de la HAS, a manifesté son intention de saisir l’ANSM afin de proposer l’élaboration d’une RTU pour Avastin dans le traitement de la DMLA.
480. Cette dépêche, relayée en interne par Novartis, reprend les déclarations suivantes de Monsieur X… : « ‘‘Il n’y a pas de raison qu'il y ait une AMM pour l'un et pas pour l'autre quand, en plus, l'un coûte beaucoup moins cher que l'autre’’, a déclaré mardi M. X…. ‘‘Nous souhaitons saisir l'ANSM, comme nous l’y autorise la loi et comme le peuvent les centres de référence pour les maladies rares, si le collège confirme ce choix, pour [lui] dire qu'il serait souhaitable de solliciter une RTU" pour Avastin dans la DMLA, a-t-il ajouté’’ » (cote 9920).
♦ Le courrier de Novartis à la HAS du 3 juillet 2012
481. En réaction à cette annonce, Novartis a adressé, le 3 juillet 2012, un courrier à M. X…, avec copie au président de l’ANSM et au président du CEPS, réaffirmant la position de l’entreprise sur l’usage d’Avastin dans la DMLA (cotes 15646 à 15651).
482. Dans ce courrier, Novartis reprend l’ensemble de ses arguments sur les études rétrospectives et de comparaison, en concluant que « les signaux de tolérance liés à l’utilisation hors AMM d’Avastin en injection intravitréenne sont donc sérieux, statistiquement significatifs dans certaines études cliniques » (cote 15649).
♦ La réponse de la HAS du 31 juillet 2012
483. Le président de la HAS, M. X…, a répondu à Novartis dans un courrier du 31 juillet 2012 (cote 3336).
484. Il a tout d’abord indiqué que la HAS avait renoncé à demander une RTU pour Avastin, en raison du cadre juridique de l’époque, qui ne réservait cette possibilité qu’au cas d’absence d’une alternative médicamenteuse appropriée.
485. Sur la question de la tolérance d’Avastin, il a toutefois précisé, en réponse aux arguments de Novartis : « Concernant la sécurité des patients recevant des injections intra-vitréennes de Bevacizumab [Avastin], nous pensons qu'il faut séparer les événements indésirables systémiques et les complications des injections locales. Pour les premiers, l'augmentation d'effets indésirables mis en évidence dans l'étude CATT ne correspond pas, comme cela est souligné par les auteurs des articles, à un effet pharmacodynamique de l'anti-VEGF. Globalement, dans les deux études randomisées, il n'y a pas de différences significatives ni en termes d'efficacité ni en termes de tolérance. Pour ce qui concerne les complications liées aux injections, notamment les cas d'endophtalmie, ils sont également décrits avec le Ranibizumab [Lucentis] et ne sont pas significativement plus fréquents dans le groupe Bevacizumab [Avastin]. Ce risque justifie les précautions d'emploi indiquées dans la recommandation publiée en 2009 par l'AFSSAPS ».
♦ La rencontre entre Novartis et les représentants du ministère de la santé d’août 2012
486. Le 6 août 2012, les représentants de Novartis ont été reçus par le ministère de la santé, comme l’indique un communiqué de l’AFP du 7 août 2012 (cote 14153).
487. Selon ce communiqué, cette rencontre a été l’occasion d’une discussion sur les « ‘conséquences à tirer’ des résultats de deux études, menées au Royaume-Uni et aux Etats Unis et publiées au printemps dernier, qui ont conclu à l'efficacité identique des deux médicaments ». Ce rendez-vous a permis à Novartis de présenter aux pouvoirs publics sa vision des études de comparaison disponibles à l’époque.
♦ Le courrier de Novartis au directeur du cabinet de la ministre des affaires sociales et de la santé du 9 août 2012
488. Dans le prolongement de cet entretien, Novartis a envoyé au directeur de cabinet de la ministre de la santé, un courrier centré sur la justification du prix de Lucentis et l’absence de légitimité de la demande de renégociation tarifaire exprimée précédemment.
489. Outre les éléments concernant directement la question de l’usage d’Avastin dans le traitement de la DMLA, Novartis évoque dans ce courrier l’importance de sa présence en France et des emplois qui y sont associés (cotes 2746 et 45098 à 45099). Le courrier souligne en particulier que les considérations liées aux coûts respectifs pour la collectivité de Lucentis et Avastin « sont posées au mépris tant des données scientifiques disponibles pour les deux spécialités, mettant en lumière des différences significatives de tolérance au détriment d’Avastin, que du cadre juridique relatif aux conditions d’autorisation de mise sur le marché (…) ».
490. Une copie de ce courrier a été envoyée, le même jour, au directeur général de la santé, M. L…. Dans son courrier d’envoi, Novartis ajoutait des éléments relatifs au changement du RCP d’Avastin : « suite aux nombreux signaux de sécurité sur l'utilisation intravitréenne d'Avastin, l’Agence Européenne du Médicament (EMA) vient de modifier le Résumé des Caractéristiques Produit d’Avastin, pour ajouter dans sa partie 4.4 ‘Mises en garde spéciales et précautions d’emploi’, l’interdiction d’utiliser Avastin en intravitréen ainsi que les événements indésirables oculaires et systémiques qui ont été observés lors de l’utilisation hors-AMM d’Avastin en intravitréen » (cote 1529).
♦ La rencontre entre Roche et les représentants du ministère de la santé au cours de l’été 2012
491. À la même époque, les représentants de Roche ont également été reçus par le ministère de la santé, comme l’indique un courrier électronique du 1er août 2012 envoyé par un employé de Roche à un représentant du siège du groupe, lui demandant de « rassurer M… » (c'est-à-dire M. M…, alors « CEO » de Genentech).
492. Ce courriel indique que la communication de l’entreprise auprès du Gouvernement a été entendue et que Mme Marisol Touraine, ministre de la santé, a indiqué, lors d’une interview à RMC, qu’il existait selon Roche un risque d’effets secondaires en cas d’utilisation d’Avastin en ophtalmologie. Il mentionne en outre que Roche prévoit de rencontrer à nouveau la ministre de la santé après l’été, pour insister à nouveau sur le « manque de sécurité » identifié dans les études CATT à un et deux ans et IVAN (cote 14688).
493. À ce sujet, le 31 juillet 2012, un représentant de Novartis a transmis à Roche un courriel relayant le changement de position de la ministre de la santé : « changement de cap depuis ses déclarations la veille => Avastin n’a pas son AMM, il ne peut donc être prescrit, sans compter que le laboratoire (Roche) a fait état d’effets secondaires » (cote 7137), ce à quoi Roche a répondu le 1er août : « Oui. Elle est en retrait, quelqu’un a dû passer par là pour lui décrypter le dossier ».
494. Un échange de courriers électroniques internes à Roche, début août, mentionne qu’« il faudra prévoir pour notre communication avec la DGS et le cabinet de la ministre, de rappeler les conclusions des études CATT. CATT2 et IVAN afin d'argumenter les points de safety et de corriger les mauvaises interprétations ». Plus loin, il est indiqué « ce position paper validé par le médical onco [oncologie] et aussi en ligne avec la position de Genentech US sur ce sujet » (cote 7135).
♦ Le plan d’action de Novartis contre l’article 45 du PFLSS pour 2013
495. Une présentation interne de Novartis, d’octobre 2012, décrit le plan d’action complet établi par l’entreprise pour lutter contre l’article 45 du PFLSS pour 2013, adopté en conseil des ministres le 10 octobre 2012, qui introduisait le principe d’une « RTU économique » (cotes 1517 à 1519).
496. Ce plan d’action incluait les actions suivantes (« immediate actions to prevent art. 45 to be voted in its current state », cote 1518) : (i) une note confidentielle pour le président de la section sociale du Conseil d’État, (ii) une lettre pour le Premier ministre et le ministre de la santé insistant sur les risques sanitaires et la possibilité d’un scandale semblable à celui du Mediator, (iii) des rencontres avec des représentants de toutes les autorités de santé, (iv) l’utilisation du LEEM comme courroie de transmission des arguments de Novartis, (v) des contacts avec les associations de médecins, les associations de patients et les principaux KOL et (vi) la préparation d’un programme clinique complet avec l’ANSM couvrant l’ensemble des indications possibles pour les anti-VEGF et pour lesquelles Lucentis n’avait pas encore d’AMM.
497. Or, comme cela ressort des développements qui suivent, Novartis a effectivement multiplié les contacts, au cours de l’automne 2012, avec les acteurs institutionnels et politiques, afin de faire en sorte que cette modification ne soit pas adoptée par le législateur.
♦ Le courrier de Novartis au directeur général de la DGS du 1er octobre 2012
498. Le 1er octobre, Novartis a adressé un courrier au directeur général de la DGS du ministère de la santé au sujet de la « sécurité sanitaire et [du] traitement des patients atteints de DMLA » (cote 1528).
499. Dans ce courrier, Novartis évoque les effets secondaires graves identifiés dans l’étude CATT pour Avastin. Plus spécifiquement, le courrier indique : « La méta-analyse des résultats à 1 an comparant Lucentis et Avastin dans le traitement de la DMLA (étude CATT menée au Etats-Unis et étude IVAN menée au Royaume-Uni) a montré une différence statistiquement significative entre Lucentis et Avastin en défaveur de ce dernier, avec un risque de survenue d’un événement systémique sévère statistiquement supérieur dans le groupe Avastin versus Lucentis ».
♦ Le courrier de Novartis au premier ministre du 5 octobre 2012
500. Le 5 octobre, un courrier reprenant un argumentaire similaire a été envoyé au Premier ministre (cotes 1525 à 1527).
501. Dans ce courrier, Novartis souligne les risques sanitaires liés à l’utilisation d’Avastin dans le traitement de la DMLA : « Autoriser par ce biais l’usage d’un médicament n’ayant pas fait l’objet de l’ensemble des études de sécurité requises et d’un plan de gestion des risques, obligatoire pour toute AMM, alors qu’il existe une alternative présentant ces garanties de sécurité, constituerait un risque majeur sur le plan sanitaire (…) » (cote 1525). Le laboratoire revient également sur les risques liés à l’utilisation d’Avastin, identifiés selon lui dans les études de comparaison et par l’EMA lors de la modification du RCP d’Avastin : « Cette polémique [sur le coût de Lucentis] est parfaitement injustifiée et ignore totalement les graves problèmes de santé publique liés à l’utilisation d’Avastin dans le traitement de la DMLA, confirmés à de nombreuses reprises (Etudes CATT et IVAN, circulaire de la DGS du 11 juillet 2012 et précision de l’agence européenne du médicament quant à l’interdiction de l’usage d’Avastin en injection oculaire intra-vitréenne) » (cote 1526).
♦ Le courrier de Novartis au président de l’ANSM du 9 octobre 2012
502. Par courrier du 9 octobre, Novartis a communiqué à l’ANSM ses motifs d’opposition au projet de « RTU économique ».
503. Dans ce courrier, le président de Novartis Pharma souligne que les études de comparaison CATT et IVAN « confirment le profil de tolérance favorable de Lucentis ». Il précise que « Ces 2 études ayant évalué la non-infériorité d’Avastin par rapport à Lucentis en terme d’efficacité ont mis en évidence une augmentation significative du risque d’événements indésirables graves pour Avastin (augmentation du risque de 30 %) » (cote 15642).
504. Novartis ajoute que « les publications récentes et les cas rapportés de diminution de l’acuité visuelle pouvant conduire à une cécité permanente ont conduit à un renforcement des précautions d’emploi d’Avastin liées à son utilisation intra-vitréenne par l’EMA. Cette dernière vient, en effet, de modifier le Résumé des Caractéristiques Produit d’Avastin suite à ces différentes études pour y ajouter une mise en garde indiquant qu’Avastin n’est pas formulé pour une utilisation intravitréenne et préciser les risques oculaires et systémiques en cas d’utilisation intravitréenne » (cote 15643).
♦ Les rencontres entre Novartis et les autorités publiques au cours du mois d’octobre 2012
505. En complément de ses échanges par courrier, Novartis a multiplié les réunions avec les autorités publiques.
506. Ainsi, le laboratoire a été reçu par la DGS et le cabinet du ministre de la santé les 11 et 12 octobre (courriel interne du 11 octobre 2012 : « documents pour rdv DG et ministère (…) veuillez trouver ci-joint les éléments pour vos rdv d’aujourd’hui et de demain », contenant en pièce jointe un document intitulé « Note Lucentis / Avastin », cote 14161 ; voir également cote 14171).
507. La « Note Lucentis / Avastin » commence par rappeler que Lucentis a été formulé spécifiquement pour une utilisation en injection intravitréenne.
508. Après avoir présenté les résultats en termes d’efficacité comparée de Lucentis et Avastin issus des études de comparaison CATT et IVAN, la note se concentre sur les données de sécurité. S’agissant de l’étude CATT, Novartis indique notamment que « le pourcentage d’évènements indésirables graves sous Avastin est plus important que sous Lucentis (39.9% vs 31.7 %; p=0.004), ce qui correspond à une augmentation du risque de 30 % (…) ». De même, la note précise que « la méta-analyse des résultats à 1 an de l’étude CATT avec ceux de l’étude IVAN confirment les résultats à un an de l’étude CATT, avec un risque de survenue d’un événement systémique sévère statistiquement supérieur dans le groupe Avastin » (cote 14162).
509. Novartis mentionne ensuite les résultats des études rétrospectives Curtis et Gower de la même façon que dans son courrier précité du 9 mai 2011 : une augmentation du risque de mortalité et d’accident vasculaire cérébral hémorragique dans Curtis, et une augmentation du risque de mortalité, d’accident vasculaire cérébral hémorragique et d’inflammation intraoculaire dans Gower (cotes 14163 et 14164).
510. Dans la suite de son argumentaire, Novartis relie ensuite ces données avec les actions récentes de certaines autorités de santé. Ainsi, toujours selon Novartis, « ces signaux sérieux de sécurité ont été reconnus par les Autorités sanitaires françaises, européennes et internationales » (cote 14164). Novartis cite notamment la prise de position de l’AFSSAPS en 2009 et en 2011, la circulaire de la DGS de juillet 2012.
511. Novartis mentionne également « l’EMA [agence européenne du médicament], qui a modifié le RCP d’Avastin pour y ajouter une mise en garde indiquant qu’Avastin n’est pas formulé pour une utilisation IVT et précise les risques oculaires et systémiques en cas d’utilisation IVT ».
512. Cet argumentaire figure également, sous une forme simplifiée, dans un document d’une page, établi spécifiquement pour le débat sur le PFLSS 2013 et le projet de « RTU économique ». Ce document reprend les mêmes arguments sur les effets indésirables d’Avastin dans les études Curtis, Gower, CATT et IVAN, ainsi que sur les actions des autorités sanitaires. Il ajoute que la possibilité d’une RTU économique pourrait conduire à une inégalité en termes de sécurité entre les patients traités à l’hôpital et ceux traités en ville (cote 14173).
513. De même, Novartis a eu un rendez-vous avec le CEPS le 12 octobre (cote 14606). La présentation associée à ce rendez-vous (cotes 14617 à 14619) revient sur le « risque sanitaire potentiel » lié à l’utilisation d’Avastin. Le laboratoire souligne en particulier que les études cliniques CATT et IVAN ont fait ressortir que « la tolérance d’Avastin est significativement inférieure à celle de Lucentis en intra-vitréenne dans la DMLA » et que « ces signaux de tolérance ont été reconnus par les Autorités sanitaires : par l’ANSM (note d’information 09/2009) ; la DGS (instruction 08/2012), l’EMA (modification du RCP d’Avastin 08/2012 (…) » (cote 14617). Enfin, le document insiste sur les « différences structurelles », les « différences pharmacologiques », les « formulations différentes » entre Avastin et Lucentis, pour en conclure que « toutes ces différences impliquent des risques en termes de tolérance locale et systémique » (cote 14619).
514. Enfin, un courrier du 16 octobre 2012 adressé au médecin conseil chef de service, responsable adjoint du département des produits de santé de la CNAMTS, reprend l’ensemble des arguments du discours établi par Novartis à cette époque afin de lutter contre la proposition faite dans le PLFSS 2013 (cotes 14257 à 14259).
♦ Le « position paper » de Roche du 17 octobre 2012
515. Dans un document interne du 17 octobre 2012, Roche expose la stratégie juridique pour contrer les projets de « RTU économique » du Gouvernement (cotes 5245 à 5249).
516. Ce document contient tout d’abord un rappel de la « dernière position officielle de Roche (courrier à l’AFSSAPS du 26 mai 2011 & 10 septembre 2012) », reprenant un argumentaire détaillé sur la moins bonne tolérance d’Avastin par rapport à Lucentis : « Les résultats des études publiées (CATT2 et Gower et al.) montrent des différences potentiellement importantes entre les profils de tolérance d’Avastin et de Lucentis dans le traitement de la DMLA : le risque d’événements indésirables graves est potentiellement plus élevé lors d’une utilisation hors AMM intra-vitréenne d’Avastin » (cote 5245).
517. Ensuite, le document revient sur les différences entre les deux molécules et présente une analyse des derniers résultats des études scientifiques publiées (cotes 5246 à 5248).
518. Enfin, le document donne la « position du juridique », consistant à contester le projet de RTU pour Avastin. Il est notamment indiqué : « Nous engagerons tous les recours en cas de RTU DMLA pour Avastin » (cote 5249).
♦ Le courriel de Novartis au représentant du LEEM du 22 octobre 2012
519. Le 22 octobre, Novartis a adressé à deux représentants du LEEM un courriel indiquant : « Pour vous permettre d’argumenter dans vos contacts, vous trouverez ci-joint des éléments, notamment de sécurité qui démontrent pourquoi l’exemple d’Avastin n’est pas fondé à appuyer la démonstration des motifs de l’article 45 », auquel est joint une note, intitulée « Note de Problématique RTU pour des raisons économiques - projet d’article 45 » (cotes 14253 à 14259).
520. Cette note reprenait l’ensemble des arguments développés par Novartis auprès des autorités publiques, exposés ci-dessus, et était accompagnée également de la lettre susvisée au médecin conseil chef de service de la CNAMTS.
♦ Les échanges de Roche avec l’ANSM à l’automne 2012
521. Par un courrier du 30 août 2012, l’ANSM a demandé à Roche de communiquer sa position après la publication des résultats à deux ans de CATT et de fournir des données supplémentaires sur Avastin en injection intraoculaire en France et en Europe (cotes 6875 à 6876 et 16428).
- Le courrier de Roche à l’ANSM du 10 septembre 2012
522. Par un courrier du 10 septembre 2012, Roche réitère sa position concernant le développement d’Avastin en ophtalmologie (cotes 5518 à 5520 et 16431 à 16433).
523. Plus spécifiquement, Roche affirme que les résultats de CATT à deux ans relèvent 30 % d’effets indésirables systémiques en plus pour Avastin. Le courrier indique également que les cas d’endophtalmies seraient plus fréquents, tout en reconnaissant que cette différence ne serait pas significative du point de vue statistique.
524. Roche mentionne ensuite le changement de RCP d’Avastin par l’EMA.
- Le courrier de Roche à l’ANSM du 17 octobre 2012
525. Par un nouveau courrier du 17 octobre 2012 (cotes 6873 et 6874), en réponse à une demande formulée par l’ANSMquelques jours auparavant concernant des données relatives à Avastin et Lucentis, Roche rappelle les différences pharmacologiques entre le ranibizumab (Lucentis) et le bevacizumab (Avastin) et en déduit que le choix de Lucentis en ophtalmologie et d’Avastin en oncologie était rationnel.
526. Le courrier conclut en indiquant que Lucentis a été spécifiquement élaboré pour un usage dans l’œil et que son profil de sécurité est bien établi, notamment en raison de son usage effectif depuis cinq ans.
- Le courrier de Roche à l’ANSM du 12 novembre 2012
527. Faisant suite à une demande orale du président de l’ANSM, Roche a adressé un courrier à l’ANSM le 12 novembre 2012 (cotes 5526 à 5532 et 16438 à 16444).
528. Dans ce courrier, Roche souligne que Lucentis a été développé spécifiquement pour une utilisation dans l’œil parce que ce produit permettrait de limiter le passage de la molécule dans le sang ainsi qu’une élimination plus rapide du principe actif et donc, de limiter les risques d’effets indésirables systémiques.
529. Roche détaille dans ce document les différences moléculaires entre Avastin et Lucentis et présente les données disponibles en termes de passage de la molécule dans le sang, qui serait plus important pour Avastin que pour Lucentis. Puis, Roche liste les résultats en termes d’effets indésirables issus de CATT et indique que, bien que l’entreprise ne puisse pas être certaine de l’existence d’un lien direct entre une exposition plus forte à Avastin après injection dans l’œil et une plus forte fréquence des effets secondaires indésirables, l’ensemble de ces données tendrait à confirmer ce raisonnement.
530. Roche continue en évoquant le changement de RCP d’Avastin. Il affirme notamment que le CHMP avait demandé l’ajout d’un paragraphe portant sur des effets indésirables systémiques potentiels après usage d’Avastin.
531. Un échange de courriels internes du 30 octobre 2012 montre que ce courrier a été rédigé en étroite collaboration avec le siège du groupe Roche (cotes 14722 à 14724).
♦ Les déclarations publiques de Roche à l’automne 2012
532. Une dépêche d’APM en date du 23 octobre 2012 reprend des déclarations du président directeur général du groupe Roche au sujet de l’utilisation d’Avastin pour le traitement de la DMLA (cotes 45117 et 7123).
533. Ce document indique : « Interrogé sur le sujet, N… [président directeur général de Roche] a souligné qu'Avastin avait été mis au point pour une utilisation systémique, avec une longue demi-vie(31), ce qui signifie aussi un risque d'effets secondaires mais qui sont acceptables dans le cadre d'un traitement du cancer. Au contraire, Lucentis, s'il a un mécanisme similaire, a été développé spécifiquement pour l'ophtalmologie, avec une demi-vie plus courte, permettant une disparition du produit en quelques jours. Le dirigeant a assuré que l'utilisation d'Avastin* dans la DMLA n'est "pas sûre". "Bien sûr c'est beaucoup moins cher, mais vous mettez en danger la santé des patients", a-t-il déclaré ».
534. Il découle d’un échange de courriels internes à Novartis du même jour que ces déclarations du directeur général de Roche ont été utilisées par Novartis dans ses contacts avec des députés français dans le cadre du débat sur le PFLSS 2013 (cote 45117).
La communication de Genentech, Roche et Novartis après l’adoption de la LFSS 2013 au cours de l’année 2013
535. Au cours de l’année 2013, après l’adoption de la réforme des RTU dans la LFSS 2013, Novartis et Roche ont poursuivi et mis à jour leur communication auprès des autorités publiques sur le sujet de l’utilisation d’Avastin dans le traitement de la DMLA.
Les déclarations publiques de Roche
536. Dans un document regroupant les éléments de langage pour une conférence de presse de M. N…, le président directeur général du groupe Roche, en France le 23 octobre 2013, Roche mentionne les effets indésirables systémiques causés par Avastin et le fait que ceux-ci sont probablement liés au passage d’Avastin dans le sang. Il réaffirme que le profil de sécurité de Lucentis est bien établi (cotes 14639 à 14640).
537. De même, un document intitulé « Briefing for M. N…'s visit to France », du mois de mars 2014 indique que Roche a réaffirmé aux autorités de santé sa position sur le manque de sécurité pour Avastin en injection intravitréenne (« we reaffirmed Roche position on the lack of safety of Avastin used as an intraocular to health authorities », cote 7824).
Le plan d’action de Novartis LFSS 2013 du 6 février 2013
538. Par un courrier électronique en date du 7 février 2013, Novartis Pharma a transmis au siège du groupe une présentation des nouvelles dispositions issues de la LFSS pour 2013, ainsi qu’un plan d’action associé (cote 3463, cotes 2877 à 2881).
539. Une grande partie des actions envisagées concernaient des recours précontentieux ou contentieux (cotes 2778 à 2880). Étaient également prévues : la mise en place d’un plan de communication (« advocacy plan ») ; la mobilisation du LEEM sur le sujet ; la rencontre de syndicats locaux de pharmaciens (au travers du LEEM) et une attention particulière au décret d’application, avec la possibilité de fournir des commentaires au travers du LEEM.
L’entretien de Novartis avec M. O…, directeur général de l’Union Nationale des Caisses d’Assurance Maladie (UNCAM)
540. Un courriel interne à Novartis du 4 septembre 2013 fait état d’une réunion prévue entre Novartis et M. O…, directeur général de l’UNCAM (cote 45090).
541. En effet, en vue des débats autour du PFLSS 2014, la CNAMTS avait proposé un certain nombre de mesures d’économie, et notamment la baisse du prix de Lucentis, ainsi que l’adoption d’une RTU pour Avastin (cotes 14200 et 14208).
542. Dans le cadre de cette rencontre, Novartis entendait notamment réagir à la proposition faite par la CNAMTS dans son rapport en vue du PFLSS 2014 de réduire significativement le prix de Lucentis (à ce sujet, cotes 14292 à 14294) : « Cette proposition est difficilement justifiable alors même que la CNAMTS reconnaît en toutes lettres que les études font apparaître ‘‘un risque plus important d’évènements indésirables graves sous bevacizumab par rapport au ranibizumab’’. La sécurité et la tolérance, tellement mis en avant suite à l’affaire Mediator et dans la dernière loi médicament, n’auraient-elles finalement pas de valeur ? […] » (cote 45090).
543. En outre, Novartis prévoyait d’utiliser, lors de cet entretien, les arguments du communiqué de presse du 14 mai 2013, qu’elle avait publié dans un contexte de publication des résultats de l’étude GEFAL, ce document figurant parmi les documents joints au courrier électronique (cote 14211).
544. L’argumentaire sur les effets indésirables d’Avastin est également repris dans une note intitulée « Impact de l’article 57 de la LFSS 2013 - Cas Lucentis / Avastin dans le traitement de la DMLA néovasculaire » (cotes 45092, 45093 et 14215), jointe au même courriel interne (cote 45090). Cette note indique notamment que : « l’EMA a ajouté dans le Résumé des Caractéristiques Produit d’Avastin une mise en garde sur les risques oculaires et systémiques en cas d’utilisation intravitréenne d’Avastin » (cote 14215).
c) Conclusion
545. Il ressort de ce qui précède que Roche et Novartis ont transmis, avec l’appui de Genentech, un argumentaire scientifique similaire concernant les risques d’utilisation d’Avastin en ophtalmologie, à l’ANSM et à d’autres acteurs institutionnels du secteur de la santé. Les laboratoires ont également mis en œuvre un certain nombre de pratiques consistant à faire pression sur les pouvoirs publics et obstacle aux procédures administratives.
F. LES GRIEFS NOTIFIES
546. Le 15 janvier 2019, les services d’instruction ont notifié les griefs suivants :
« Grief n° 1
Il est fait grief :
- à la société Novartis Pharma S.A.S. (RCS 410 349 070), en tant qu’auteur des pratiques,
- à la société Novartis AG, en tant qu’auteur des pratiques,
- aux sociétés Novartis Groupe France S.A. (RCS 709 804 538) et Novartis AG, en tant que sociétés mères de Novartis Pharma S.A.S.,
d’avoir abusé de la position dominante détenue collectivement avec Roche et Genentech sur le marché français du traitement de la DMLA exsudative par anti-VEGF, en élaborant et en exécutant une stratégie visant à limiter le recours par les médecins ophtalmologistes au produit Avastin, concurrent de son médicament Lucentis pour ce type de pathologies, au travers de différentes actions de communication à caractère trompeur vis-à-vis des professionnels de santé, des autorités du secteur de la santé, des patients et du grand public.
Cet ensemble de comportements, ayant débuté le 10 mars 2008 et ayant pris fin à tout le moins au mois de novembre 2013, constitue une pratique prohibée par les articles 102 du TFUE et L. 420-2 du code de commerce.
Grief n° 2
Il est fait grief :
- à la société Novartis Pharma S.A.S. (RCS 410 349 070), en tant qu’auteur des pratiques,
- à la société Novartis AG, en tant qu’auteur des pratiques,
- aux sociétés Novartis Groupe France S.A. (RCS 709 804 538) et Novartis AG, en tant que sociétés mères de Novartis Pharma S.A.S.,
- à la société Roche (RCS 552 012 031), en tant qu’auteur des pratiques
- à la société Genentech, Inc. en tant qu’auteur des pratiques,
- à la société Roche Holding AG, en tant que société mère de Roche et Genentech, Inc.,
d’avoir abusé de la position dominante qu’elles détiennent collectivement sur le marché français du traitement de la DMLA exsudative par anti-VEGF, en élaborant et en exécutant une stratégie visant à faire obstacles aux initiatives des pouvoirs publics français permettant d’autoriser administrativement l’usage d’Avastin pour le traitement de la DMLA, au travers d’un ensemble de comportements de blocage administratif et de diffusion d’un discours trompeur aux responsables politiques et aux autorités de santé.
Cet ensemble de comportements, ayant débuté le 7 avril 2008 et ayant pris fin à tout le moins au mois de novembre 2013, constitue une pratique prohibée par les articles 102 du TFUE et L. 420-2 du code de commerce ».
II. Discussion
547. Seront successivement examinés la compétence de l’Autorité (A), la procédure (B), l’applicabilité du droit de l’Union (C), le bien-fondé des griefs notifiés (D), l’imputabilité (E) et les sanctions (F).
A. SUR LA COMPETENCE
548. Les sociétés mises en cause soutiennent que l’Autorité n’est pas compétente pour analyser la pertinence de leurs échanges avec les autorités de santé. Elles font valoir que l’Autorité ne dispose pas de l’expertise nécessaire pour porter une appréciation critique sur la valeur des arguments scientifiques présentés par Roche et Novartis à leurs divers interlocuteurs, dans un contexte d’incertitude scientifique sur l’efficacité et la sécurité de l’utilisation d’Avastin dans le traitement de la DMLA. Roche et Novartis soutiennent en outre que l’Autorité ne peut se prononcer sur la légalité des décisions prises par les autorités de santé dans l’exercice de leurs prérogatives de puissance publique.
549. Toutefois, l’Autorité est compétente pour connaître de toute pratique susceptible de constituer une infraction aux règles de concurrence, et ce quel que soit le secteur d’activité concerné ou le vecteur utilisé (décision n° 17-D-25 du 20 décembre 2017 relative à des pratiques mises en œuvre dans le secteur des dispositifs transdermiques de fentanyl, points 354 à 358, confirmée par un arrêt de la cour d’appel de Paris du 11 juillet 2019, société Janssen-Cilag SAS, n° 18/01945). Il lui appartient à cet effet de procéder elle-même à une analyse du contexte juridique et factuel dans lequel s’inscrivent les pratiques litigieuses. À défaut, elle serait « captive d’une analyse du contexte qui pourrait avoir été retenue par une autre autorité administrative à la suite d’un comportement susceptible d’être anticoncurrentiel » (arrêt de la cour d’appel de Paris du 11 juillet 2019, société Janssen-Cilag SAS, n° 18/01945, point 98).
550. Lorsque les pratiques sont en lien avec la commercialisation de médicaments, l’Autorité tient compte du cadre juridique applicable, notamment en ce qui concerne la procédure de délivrance des AMM au niveau européen et au niveau national (décision n° 17-D-25 du 20 décembre 2017 relative à des pratiques mises en œuvre dans le secteur des dispositifs transdermiques de fentanyl, point 356). A cet égard, contrairement à ce que soutiennent les parties, la jurisprudence européenne, et notamment l’arrêt de la Cour de justice du 23 janvier 2018 rendu sur une question préjudicielle dans l’affaire italienne Roche/Novartis (arrêt Hoffmann-Laroche, C-179/16), n’a jamais écarté la compétence d’une autorité nationale de concurrence pour procéder à une telle analyse.
551. Sans se substituer aux autorités scientifiques, ni statuer sur la légalité des décisions prises par les autorités de santé, il revient à l’Autorité de qualifier les pratiques dont elle est saisie sous l’angle du droit de la concurrence. Dans ce cadre, il lui incombe d’analyser le comportement des entreprises mises en cause sur un marché donné. Les actions des laboratoires pharmaceutiques auprès des autorités de santé relèvent ainsi de sa compétence lorsqu’elles sont susceptibles de s’inscrire dans une stratégie anticoncurrentielle. Tel est notamment le cas s’agissant de pratiques visant à entraver la commercialisation de médicaments concurrents (avis n° 13-A-24 du 19 décembre 2013 relatif au fonctionnement de la concurrence dans le secteur de la distribution du médicament à usage humain en ville, point 423).
552. En l’espèce, comme il sera démontré aux paragraphes 760 et suivants de la présente décision, les pratiques mises en œuvre par l’entité collective constituée par Roche, Novartis et Genentech afin de faire échec à l’utilisation d’Avastin en ophtalmologie sont examinées par l’Autorité, en ce qu’elles sont de nature à influencer la structure du marché en contribuant à favoriser l’usage d’un médicament (Lucentis) au détriment d’un produit concurrent (Avastin).
553. S’il est exact que la qualification de ces pratiques nécessite d’examiner le contenu du discours véhiculé par l’entité collective sur les risques liés à l’utilisation d’Avastin « hors AMM » en ophtalmologie, une telle analyse n’implique pas que l’Autorité se prononce sur la validité des arguments scientifiques opposés dans le cadre du débat public sur l’efficacité et la sécurité comparées d’Avastin et Lucentis. Elle consiste uniquement à déterminer si Roche et/ou Novartis ont reproduit fidèlement ces arguments, ou les ont dénaturés par une présentation dépourvue de la mesure et de l’objectivité requise, compte tenu des incertitudes du débat public. Par ailleurs, l’Autorité n’est pas davantage conduite à se prononcer sur la légalité des décisions prises par les autorités de santé. À cet égard, si Novartis soutient que l’Autorité ne démontre pas l’existence de comportements « détachables de l’appréciation de la légalité de décisions administratives » (observations de Novartis, cote 49191), cet argument doit être écarté, dès lors que les griefs notifiés ne visent pas les décisions ou l’action des autorités de santé, mais le comportement des membres de l’entité collective composée par Roche, Novartis et Genentech.
554. Ainsi, contrairement à ce que soutiennent les sociétés mises en cause, l’analyse de l’Autorité repose sur la qualification juridique des pratiques en cause au regard du droit de la concurrence. Elle consiste à déterminer si, dans le cadre de leurs relations avec les professionnels de santé, les associations de patients, le grand public et les autorités publiques, les entreprises mises en cause ont eu recours à des moyens étrangers à une concurrence par les mérites pour favoriser la commercialisation d’un de leurs produits.
555. Il en résulte que les sociétés mises en cause ne sont pas fondées à soutenir que l’Autorité ne serait pas compétente pour analyser, qualifier et, le cas échéant, sanctionner, les pratiques en cause.
B. SUR LA PROCEDURE
1. CONCERNANT LA DUREE DE LA PROCEDURE
556. Roche et Genentech soutiennent que la longueur inhabituelle de la procédure, résultant notamment de la période de plus de six ans et demi qui s’est écoulée entre le premier signalement reçu par la DGCCRF le 31 janvier 2012 et l’envoi de la notification de griefs le 23 janvier 2019, a entraîné une violation de leurs droits de la défense.
557. Mais il résulte d’une jurisprudence constante que l’exigence relative au délai raisonnable de la procédure, qui découle de l’article 6, paragraphe 1, de la CEDH, doit être appréciée au regard de l’ampleur et de la complexité de l’affaire (voir notamment cour d’appel de Paris, 26 janvier 2012, Beauté Prestige International, n° 2010/23945).
558. Par ailleurs, il sera rappelé à toutes fins utiles que la sanction qui s’attache à la violation éventuelle par l’Autorité de l’obligation de se prononcer dans un délai raisonnable n’est pas l’annulation de la procédure, mais la réparation du préjudice résultant éventuellement du délai excessif (arrêt de la cour d’appel de Paris du 21 décembre 2017, La Banque Postale e.a., n° 15/17638). Il n’en va autrement que s’il est démontré une atteinte personnelle, effective et irrémédiable au droit des entreprises en cause de se défendre (même arrêt). C’est à l’entreprise qui soutient que la durée excessive de la procédure a fait obstacle, concrètement et effectivement, à l’exercice normal de ses droits de la défense, d’en apporter la preuve (arrêt de la cour d’appel de Paris du 18 janvier 2018, Groupement des installateurs français SA, n° 2017/01703).
559. En ce qui concerne les atteintes alléguées à la possibilité pour les entreprises de se défendre utilement contre les griefs notifiés compte tenu de la durée de la procédure, la cour d’appel de Paris a précisé que « la réalité d’une telle violation s’apprécie nécessairement à l’aune du devoir général de prudence incombant à chaque opérateur économique qui se doit de veiller à la bonne conservation de ses livres et archives comme de tous éléments permettant de retracer la licéité de ses pratiques en cas d’actions judiciaire ou administrative » (arrêt de la cour d’appel de Paris du 26 janvier 2012, Beauté Prestige International, n° 2010/23 945).
560. En l’espèce, la durée de l’instruction doit être appréciée au regard de la complexité de l’affaire, qui a nécessité une analyse approfondie d’un secteur étroitement réglementé, la compréhension des pratiques impliquant une connaissance fine des règles d’autorisation de mise sur le marché et de détermination du prix des médicaments, du rôle respectif des différentes autorités de santé, des relations entre les laboratoires pharmaceutiques et les médecins, ou encore des prises de position des autorités de santé, françaises et européenne, sur ces questions. En outre, au plan procédural, l’affaire a donné lieu à des opérations de visite et de saisie, au cours desquelles un nombre important de données a été saisi, et qui ont été contestées devant les juridictions compétentes (cf. paragraphe 2 de la présente décision).
561. Ainsi, compte tenu de l’ampleur et de la complexité de l’affaire, la durée de l’instruction ne saurait, dans les circonstances de l’espèce, être considérée comme ayant méconnu la garantie du délai raisonnable.
562. En tout état de cause, Roche et Genentech ne démontrent pas que la durée de la procédure aurait fait obstacle à l’exercice effectif de leurs droits de la défense. Ces sociétés ne sauraient utilement invoquer à ce titre, de façon générale, la « lenteur des mesures d’investigation ». Contrairement à ce qui est allégué, et compte tenu des actes réalisés au cours de l’instruction, l’écoulement du temps n’a pas rendu impossible la collecte et la conservation des éléments de preuve contemporains aux pratiques en cause. À cet égard, la notification de griefs et le rapport exposent clairement, documents à l’appui, l’évolution du contexte d’incertitude concernant l’utilisation d’Avastin pour le traitement de la DMLA, et les prises de position des différentes autorités de santé. Par ailleurs, si les auditions de certains représentants des autorités de santé et des praticiens ont été menées plusieurs années après que les pratiques ont cessé, cette circonstance – qui est inhérente à la conduite de la procédure d’instruction – ne saurait caractériser par elle-même une atteinte aux droits de la défense des entreprises mises en cause.
563. Enfin, Roche et Genentech, qui ont pu avoir connaissance des investigations menées par l’Autorité au plus tard le 8 avril 2014, date des opérations de visite et de saisie, indiquent elles-mêmes ne pas avoir rencontré de difficultés pour accéder aux documents utiles à leur défense. Elles ont d’ailleurs été en mesure de présenter des observations détaillées en réponse à la notification de griefs et au rapport qui leur ont été notifiés.
564. Il en résulte qu’aucune atteinte aux droits de la défense liée à la durée de la procédure n’est établie en l’espèce.
2. CONCERNANT LA REUNION AVEC DES REPRESENTANTS DE LA HAS
565. Roche et Genentech soutiennent que le principe du contradictoire a été méconnu dès lors que la réunion qui s’est tenue entre les services d’instruction et des représentants de la HAS le 18 janvier 2016 n’a pas donné lieu à l’établissement d’un procès-verbal versé au dossier.
566. Il résulte de l’article L. 450-2 du code de commerce que « les enquêtes donnent lieu à l’établissement de procès-verbaux et, le cas échéant, de rapports (…) ». L’article R. 463-6 du même code dispose que « les auditions auxquelles procède le rapporteur donnent lieu à un procès-verbal, signé par les personnes entendues. En cas de refus de signer, il en est fait mention par le rapporteur. Les personnes entendues peuvent être assistées d’un conseil ».
567. D’abord, il convient de relever que si l’article R. 463-6 du code de commerce prévoit de façon générale que toute audition doit donner lieu à l’établissement d’un procès-verbal, l’absence d’un procès-verbal n’est pas, par elle-même, de nature à entacher la régularité de la procédure suivie à l’égard des entreprises mises en cause. Dans une telle configuration, l’incidence de l’absence de procès-verbal doit être appréciée au cas par cas, au regard des principes qui gouvernent le respect des droits de la défense et des garanties dont doivent bénéficier les entreprises mises en cause.
568. À cet égard, la Cour de justice a précisé, s’agissant de l’absence de communication de documents prétendument à décharge, qu’il incombe non seulement à l’entreprise d’établir qu’elle n’a pas eu accès à ces éléments de preuve, mais également qu’elle aurait pu les utiliser pour sa défense (voir les arrêts de la Cour de justice du 1er juillet 2010, Knauf Gips KG, aff. C-407/08 et du 6 septembre 2017, Intel Corporation Inc., aff. C-413/14).
569. Ensuite, à supposer que l’absence de procès-verbal constitue une irrégularité de procédure, il résulte de la jurisprudence du Conseil d’État qu’un vice affectant une procédure menée par une autorité administrative, lié notamment à la consultation d’un organisme, n’est de nature à entacher d’illégalité la décision prise à l’issue de cette procédure que si ce vice a été susceptible d’exercer une influence sur le sens de la décision ou s’il a privé les intéressés d’une garantie (décision d’Assemblée du 23 décembre 2011, Danthony, n° 335033, publiée au recueil, dont les principes ont ensuite été codifiés au code des relations du public avec l’administration)(32).
570. Enfin, la jurisprudence n’exige pas que les éléments de preuve collectés au cours de l’enquête soient versés au dossier sous une forme particulière, dès lors que les informations recueillies sont soumises au débat contradictoire et que les entreprises mises en cause sont en mesure d’en prendre connaissance et d’en discuter (arrêt de la cour d’appel de Paris du 21 décembre 2017, La Banque Postale e.a., n° 15/17638).
571. En l’espèce, à la suite de l’envoi de la notification de griefs, Roche et Genentech ont, par l’intermédiaire de leur conseil, sollicité le 5 février 2019 la communication de plusieurs documents et notamment du procès-verbal de l’audition des représentants de la HAS ayant eu lieu le 18 janvier 2016 (cotes 47394 et 47395). Par courrier électronique du 7 février 2019, le rapporteur précisait que la réunion du 18 janvier 2016, qui ne portait que sur des éléments déjà fournis par la HAS, n’avait donné lieu à aucun procès-verbal (cote 47397).
572. S’agissant d’abord des conditions dans lesquelles cette réunion s’est tenue, il importe de préciser qu’elle a été précédée d’échanges, sous forme écrite, entre les services d’instruction de l’Autorité et la HAS. Le 9 décembre 2015, plusieurs publications de la HAS(33) concernant le traitement de la DMLA ont ainsi été transmises au rapporteur alors en charge de l’affaire par le conseiller du président et du directeur de la HAS (cotes 13197 et suivantes). Par courrier électronique du 14 décembre 2015, le rapporteur a accusé réception de ces documents et adressé à son interlocuteur un questionnaire portant sur le traitement de la DMLA et sur deux documents spécifiques : d’une part, un courrier adressé le 31 juillet 2012 à Novartis par le président de la HAS et, d’autre part, la circulaire de la direction générale de la santé en date du 11 juillet 2012 (cotes 13450 et suivantes). Par courrier électronique du 13 janvier 2016, le conseiller auprès du président et du directeur de la HAS a fait parvenir au rapporteur des documents complémentaires(34) ainsi que les réponses au questionnaire, en précisant notamment que la réunion du 18 janvier 2016 pourrait permettre d’évoquer ce questionnaire (cotes 13517 et suivantes). Tous ces échanges de courriers électroniques, ainsi que l’ensemble des documents qui y étaient annexés, figuraient au dossier auquel les entreprises mises en cause ont eu accès.
573. Ensuite, il convient de relever que, ni la notification de griefs, ni le rapport, ne s’appuient sur les déclarations des représentants de la HAS rencontrés le 18 janvier 2016. Pour étayer le grief notifié à Roche et Genentech, les services d’instruction se sont exclusivement fondés sur des documents auxquels les entreprises mises en cause ont pu accéder, et qu’elles ont été en mesure de commenter, dans le cadre de la procédure contradictoire.
574. Enfin, les entreprises mises en cause ne peuvent être regardées, dans les circonstances de l’espèce, comme ayant été privées d’éléments utiles à leur défense du fait de l’absence de procès-verbal de la réunion du 18 janvier 2016.
575. D’une part, Roche et Genentech soulignent qu’en l’absence de procès-verbal, il ne peut être exigé d’elles qu’elles apportent la preuve que les déclarations des représentants de la HAS lors de la réunion du 18 janvier 2016 auraient pu être utiles à leur défense. Mais il demeure qu’aucune déclaration effectuée lors de cette réunion n’a été mentionnée dans la notification de griefs. Et au demeurant, il sera relevé en tout état de cause, que les sociétés mises en cause n’ont pas demandé que les représentants de la HAS présents lors de la réunion, dont l’identité était mentionnée dans le courrier électronique du 9 décembre 2015 (cotes 13197 et suivantes), soient entendus au cours de la séance de l’Autorité. Elles n’allèguent d’ailleurs pas avoir cherché à contacter les représentants de la HAS afin de connaître la teneur des échanges intervenus lors de cette réunion. De telles circonstances figurent parmi celles que la Cour de justice a relevées, dans l’affaire Intel précitée, pour juger que la requérante n’apportait pas la preuve qu’elle aurait pu utiliser pour sa défense des éléments prétendument à décharge auxquels elle n’aurait pu avoir accès en temps utile (arrêt Intel précité, paragraphe 101).
576. D’autre part, comme indiqué aux paragraphes précédents, les échanges de courriers électroniques entre les services d’instruction et la HAS attestent de ce que la réunion du 18 janvier 2016 a été précédée de la communication de plusieurs documents auxquels les entreprises mises en cause ont pu avoir accès. Ces échanges de courriers électroniques révèlent en outre que la réunion n’a eu d’autre objet que d’évoquer et de commenter ces documents. Aucun élément ne permet de considérer que les représentants de la HAS auraient, au cours de la réunion, exprimé une position différente de celle figurant dans les documents échangés avant la réunion, et notamment dans la réponse au questionnaire adressée au rapporteur le 13 janvier 2016. Aucun élément du dossier n’indique ainsi que des éléments qui auraient pu être invoqués à décharge par les sociétés mises en cause auraient été omis du dossier.
577. Par suite, dans les circonstances de l’espèce, l’absence de réalisation d’un procès-verbal de la réunion du 18 janvier 2016 n’a privé Roche et Genentech d’aucune garantie et n’a pas été de nature à porter atteinte à l’exercice effectif des droits de la défense.
3. CONCERNANT LES ERREURS MATERIELLES AFFECTANT LA NOTIFICATION DE GRIEFS
578. Roche et Genentech exposent que la notification de griefs comporte des erreurs matérielles, concernant notamment certaines références aux cotes du dossier. Les sociétés allèguent que ces erreurs ont rendu plus complexe la lecture du dossier et ont ainsi porté atteinte à l’exercice effectif des droits de la défense.
579. La portée des insuffisances affectant la notification de griefs et ses annexes doit être appréciée en fonction de leur nature et de leur incidence concrète sur l’exercice des droits de la défense. La cour d’appel de Paris juge ainsi que si certaines pièces du dossier sont incomplètes ou difficilement lisibles, de telles insuffisances seraient de nature à réduire ou à anéantir leur force probante et non à porter atteinte au principe même de la contradiction (arrêt de la cour d’appel de Paris du 24 juin 2008, société France Travaux, n° 2006/06913).
580. En l’espèce, les erreurs invoquées par Roche et Genentech, qui concernent seulement six paragraphes (sur plus de 700) de la notification de griefs selon l’annexe 14 au mémoire présenté par ces sociétés, portent exclusivement sur des références à des cotes erronées. De telles erreurs, au demeurant circonscrites et de portée limitée – notamment compte tenu de l’ampleur du dossier et du nombre de cotes, et que, de surcroît, les mises en cause ont pu rectifier d’elles-mêmes dans leurs écritures – n’ont pas eu d’incidence sur l’exercice effectif des droits de la défense des sociétés mises en cause, qui ont été en mesure de se référer aux documents adéquats et de présenter leurs observations dans le délai imparti.
581. Il en résulte que ces erreurs ne peuvent être regardées comme ayant empêché l’exercice effectif des droits de la défense.
4. CONCERNANT L’IMPARTIALITE ET LA LOYAUTE DE LA PROCEDURE D’INSTRUCTION
582. Novartis estime que le rapport comporte de nombreuses contradictions qui démontreraient le manque de rigueur et d’objectivité de l’instruction. Il allègue en outre que l’instruction aurait été menée de façon « incomplète » et « à charge ». De même, Roche fait valoir que le rapporteur aurait manqué à son devoir d’impartialité et méconnu le droit à un procès équitable, en lui opposant des documents émanant de Novartis.
583. Il résulte de la jurisprudence de la cour d’appel de Paris que « l’appréciation de la partialité de l’instruction ne saurait résulter de la seule circonstance alléguée au cas d’espèce que le rapporteur n’aurait pas tenu compte, dans sa notification de griefs, d’éléments qui, selon les parties, viendraient au soutien de leur défense » (arrêt de la cour d’appel de Paris du 20 janvier 2011, société Perrigault, n° 2010/08165). La cour d’appel juge en outre qu’il ne peut être « reproché aux rapporteurs d’avoir retenu des éléments à charge des entreprises et écarté les éléments que celles-ci invoquaient à leur décharge, dès lors qu’ils ont pour fonction d’instruire et de décrire dans la notification de griefs, puis dans le rapport, ce qui à leurs yeux doit conduire à la qualification et à la sanction de pratiques anticoncurrentielles » (arrêt de la cour d’appel de Paris du 17 mai 2018, société Umicore, n° 2016/16621).
584. Dès lors, si Novartis allègue en l’espèce que, dans son rapport, le rapporteur aurait écarté les objections opposées à la notification de griefs et aurait omis de répondre à certains arguments, ces circonstances ne sauraient révéler une méconnaissance de l’obligation d’impartialité et un manque de loyauté dans la conduite de l’instruction.
585. Par ailleurs, si l’interprétation de certains documents retenue dans le rapport est contestée par Roche et Genentech, les mises en cause n’apportent aucun élément de nature à démontrer que le rapporteur aurait procédé à une présentation tronquée ou fallacieuse de ces documents, en méconnaissance de l’obligation d’impartialité.
586. De plus, si Roche et Genentech soutiennent que des pièces émanant de Novartis ont été utilisées pour étayer le grief qui leur a été notifié, cette circonstance ne peut par elle-même révéler une atteinte au droit à un procès équitable, dès lors que ces pièces figuraient au dossier et que les sociétés étaient en mesure d’en discuter la teneur, la portée ou la valeur probante, s’agissant des pratiques qui leur sont imputées. Si les sociétés se réfèrent en particulier à la cote 4319 citée au point 721 du rapport, elles indiquent elles-mêmes que cette pièce a été déclassée à leur demande. Elles ne sont dès lors pas fondées à alléguer une méconnaissance du droit à un procès équitable.
587. Enfin, en dénonçant les prétendues contradictions dont serait entaché le rapport, Novartis entend en réalité critiquer la pertinence du raisonnement suivi par le rapporteur, sans démontrer un quelconque manque de rigueur et d’objectivité de la procédure d’instruction.
5. CONCERNANT LA MODIFICATION ALLEGUEE DES GRIEFS NOTIFIES
588. Novartis soutient que les services d’instruction auraient, dans le rapport, fait évoluer les accusations portées contre les entreprises mises en cause dans la notification de griefs, concernant notamment le choix de Genentech de ne pas développer Avastin en ophtalmologie ou encore l’impact de sa communication sur les négociations de prix de Lucentis. Selon Novartis, cette « accusation évolutive » l’aurait empêchée de se défendre correctement.
589. Il résulte toutefois du caractère contradictoire de la procédure que l’analyse faite dans le rapport peut évoluer par rapport à celle développée dans la notification des griefs (voir, à cet égard, la décision du Conseil de la concurrence n° 07-D-23 du 12 juillet 2007, point 52). En effet, « tant que le rapport ne vise pas des pratiques différentes de celles évoquées dans la notification des griefs et ne modifie pas leur qualification, il est possible d’y affiner l’analyse concurrentielle, d’étayer ou de préciser l’un des griefs notifiés » (décision n° 06-D-18 du 28 juin 2006 relative à des pratiques mises en œuvre dans le secteur de la publicité cinématographique, point 111). La cour d’appel de Paris considère de même que « le débat contradictoire qui s’ouvre dès la notification de griefs aux parties se poursuit tout au long de la procédure et autorise les services d’instruction à faire évoluer leur analyse pour répondre aux arguments des parties » (arrêt de la cour d’appel de Paris du 21 décembre 2017, La Banque Postale e.a., n° 15/17638, point 138).
590. En l’espèce, le rapport ne vise pas des pratiques différentes de celles évoquées dans la notification de griefs. De même, la qualification des griefs est demeurée inchangée. Il était, dans ces conditions, loisible aux services d’instruction d’affiner et de compléter leur analyse afin de répondre aux observations des parties. Enfin, si Novartis soutient que les arguments successivement développés par les services d’instruction seraient infondés, voire contradictoires, elle n’expose pas en quoi les droits de la défense auraient, de ce fait, été méconnus. Ces arguments visent en réalité à contester les griefs notifiés sur le fond. Il y sera répondu infra.
6. CONCERNANT L’ETENDUE DES GRIEFS NOTIFIES
591. Roche, Genentech et Novartis soutiennent que l’étendue des griefs notifiés n’est pas claire. Plus spécifiquement, les mises en cause affirment que les griefs notifiés doivent être considérés comme portant uniquement sur le marché français du traitement de la DMLA exsudative par anti-VEGF, à l’exclusion des autres marchés du traitement des pathologies oculaires (tels ceux de l’OMD, de l’OBVR ou de l’OVCR) qui n’ont été pris en compte ni pour qualifier la position dominante collective, ni les pratiques en cause, alors même que les services d’instruction ont pris soin de les définir.
592. Toutefois, les griefs notifiés sont parfaitement clairs et cohérents avec la caractérisation des pratiques en cause.
593. Selon la pratique décisionnelle et la jurisprudence françaises, la notification de griefs doit informer les parties des pratiques reprochées, de leur qualification juridique au regard du droit applicable - national ou de l’Union - et des personnes auxquelles elles sont imputées, afin de les mettre en mesure de contester utilement, au cours de la procédure contradictoire, soit la réalité des faits, soit leur qualification, soit leur imputation. En ce sens la cour d’appel de Paris a jugé : « Considérant qu’il résulte des principes du contradictoire et du respect des droits de la défense, inscrits dans la CESDH et rappelés à l'article L. 463-1 du code de commerce aux termes duquel l'instruction et la procédure devant le Conseil sont pleinement contradictoires, comme la jurisprudence nationale et communautaire, que la communication des griefs visée en droit interne par l'article L. 463-2 du code de commerce doit contenir un exposé des griefs libellé dans des termes suffisamment clairs, fussent-ils sommaires, pour permettre aux intéressés de prendre effectivement connaissance des comportements qui leur sont reprochés par l'autorité de la concurrence (...) » (arrêt de la cour d’appel de Paris, 26 septembre 2006, n° 2005/24285, page 6 ; voir également la décision n° 05-D-64 du 25 novembre 2005 relative à des pratiques mises en œuvre sur le marché des palaces parisiens, paragraphe 113).
594. Dans l’affaire Expedia, le Conseil de la concurrence a souligné que « la notification des griefs est un document synthétique qui contient une description précise des faits reprochés, leur date, leur imputabilité et leur qualification, puis reprend, in fine, en les résumant, la rédaction des griefs eux-mêmes dans une formule concise. Elle constitue l’acte d’accusation et doit donc être précise (cour d’appel de Paris, 29 mars 2005, Filmdis Cinésogar), cette exigence n’excluant pas que les juges d’appel et de cassation recherchent, dans le corps même de la notification des griefs, la portée de ces derniers (Cour de cassation, 6 avril 1999, ODA) » (Décision n° 09-D-06 du 5 février 2009 relative à des pratiques mises en œuvre par la SNCF et Expedia Inc. dans le secteur de la vente de voyages en ligne, paragraphe 73).
595. Or, en l’espèce, si les griefs notifiés mentionnent l’existence d’un abus de position dominante collective composée par Novartis, Roche et Genentech sur le seul marché français du traitement de la DMLA exsudative par anti-VEGF, les développements de la notification de griefs éclairent en revanche les parties sur leur portée et, plus particulièrement, sur le fait qu’ils ont été susceptibles d’avoir des effets sur les marchés connexes du traitement des autres pathologies oculaires.
596. En effet, en premier lieu, la notification de griefs définit les marchés connexes du traitement des autres indications oculaires (OMB, OBVR ou OCVR), et précise que, pour ces trois marchés, Lucentis et Avastin se trouvaient en situation de concurrence (cotes 47243 et 47244).
597. En second lieu, de nombreuses pièces exploitées dans la notification de griefs montrent que Novartis s’inquiétait également de l’utilisation particulièrement importante d’Avastin pour d’autres indications en ophtalmologie que celle du traitement de la DMLA, utilisation confirmée par de nombreux médecins (cotes 7090, 7095 ; 13585 et 16589). En effet, les pratiques en cause visaient précisément à bloquer l’utilisation d’Avastin dans ces autres indications, dans l’attente d’une AMM de Lucentis. Ainsi, un document interne de Novartis montre que, pour le CHU de Besançon et le CH de Vesoul, l’objectif du laboratoire était de « s’opposer au référencement d’Avastin en ophtalmo pour indications OMD et OVR » et de « transférer les unités d’Avastin utilisé dans ces indications en unités de Lucentis » (cotes 15696 à 15700).
598. Par ailleurs, toutes ces pathologies rétiniennes traitées par injections dans l’œil d’anti-VEGF sont concernées par le débat sur la comparaison des effets secondaires indésirables entre Avastin et Lucentis. Ainsi, dès lors que la question de la tolérance et des effets indésirables est indifférente à la pathologie exacte pour laquelle ces produits sont utilisés (DMLA ou autres), leurs volumes de vente ont pu être affectés de la même manière par les comportements des mises en cause.
599. Par conséquent, l’argument sera écarté.
C. SUR L’APPLICABILITE DU DROIT DE L’UNION
1. PRINCIPES APPLICABLES
600. L’article 102 du TFUE dispose qu’« est incompatible avec le marché commun et interdit, dans la mesure où le commerce entre États membres est susceptible d’en être affecté, le fait pour une ou plusieurs entreprises d’exploiter de façon abusive une position dominante sur le marché commun ou dans une partie substantielle de celui-ci ».
601. Selon la jurisprudence de la Cour de justice et la communication de la Commission européenne portant lignes directrices relatives à la notion d’affectation du commerce figurant aux articles 101 et 102 du TFUE(35), trois éléments doivent être établis pour que des pratiques soient susceptibles d’affecter sensiblement le commerce entre États membres de l’Union : l’existence d’un courant d’échanges entre les États membres portant sur les produits en cause, l’existence de pratiques susceptibles d’affecter ces échanges et le caractère sensible de cette affectation.
602. La Commission européenne rappelle au point 19 de ses lignes directrices que la notion de commerce entre États membres n’est pas limitée aux échanges transfrontaliers traditionnels de produits et de services. Elle englobe aussi « les cas où des accords et pratiques affectent la structure de la concurrence sur le marché » (point 20).
603. Par ailleurs, la circonstance que les pratiques sanctionnées ne soient commises que sur le territoire d’un seul État membre ne fait pas obstacle à ce que le commerce entre États membres soit susceptible d’être affecté. À cet égard, la Cour de cassation a jugé, dans un arrêt du 31 janvier 2012, que les termes « susceptibles d’affecter » énoncés par les articles 101 et 102 du TFUE « supposent que l’accord ou la pratique abusive en cause permette, sur la base d’un ensemble d’éléments objectifs de droit ou de fait, d’envisager avec un degré de probabilité suffisant qu’il puisse exercer une influence directe ou indirecte, actuelle ou potentielle, sur les courants d’échanges entre États membres, sans que soit exigée la constatation d’un effet réalisé sur le commerce intracommunautaire » (arrêt de la Cour de cassation du 31 janvier 2012, Orange Caraïbe e.a., n° 10-25.772, page 6).
604. S’agissant du troisième élément, la Cour de cassation a jugé dans ce même arrêt que « le caractère sensible de l’affectation directe ou indirecte, potentielle ou actuelle, du commerce intracommunautaire résulte d’un ensemble de critères, parmi lesquels la nature des pratiques, la nature des produits concernés et la position de marché des entreprises en cause » (arrêt de la Cour de cassation du 31 janvier 2012, Orange Caraïbe e.a., n° 10-25.772, page 6 ; voir également, en ce sens, arrêt de la cour d’appel de Paris, du 28 mars 2013, Société des pétroles Shell e. a., n° 2011/18 245 et arrêt de la Cour de cassation du 20 janvier 2015, Société Chevron Products Company e. a., n° 13-16.745).
605. Enfin, la Commission européenne rappelle au point 45 de ses lignes directrices que : « L'appréciation du caractère sensible dépend des circonstances de chaque espèce, et notamment de la nature de l'accord ou de la pratique, de la nature des produits concernés et de la position de marché des entreprises en cause. Si les accords ou les pratiques sont, par leur nature même, susceptibles d'affecter le commerce entre États membres, le seuil du caractère sensible est inférieur à celui des accords et pratiques qui ne sont pas, par leur nature même, susceptibles d'affecter ce commerce. Plus la position de marché des entreprises en cause est forte, plus il est probable qu'un accord ou une pratique susceptible d'affecter le commerce entre États membres pourra être considéré comme le faisant de façon sensible ».
2. APPRECIATION EN L’ESPECE
606. Roche et Genentech soutiennent que le droit de l’Union ne serait pas applicable à la présente affaire, compte tenu, d’une part, des spécificités nationales du secteur pharmaceutique et, d’autre part, des spécificités des pratiques en cause, qui relèvent de la catégorie des abus d’exploitation, pour lesquels la caractérisation d’une affectation du commerce entre États membres est « plus complexe ».
607. Toutefois, les arguments de Roche et Genentech doivent être écartés pour les raisons suivantes, qui démontrent que les pratiques en cause étaient de nature à dépasser les frontières nationales et affecter le commerce des États membres.
608. En premier lieu, nonobstant les particularités du cadre réglementaire propre à chaque État et les spécificités nationales liées à l’usage plus ou moins répandu d’Avastin dans le traitement de la DMLA, les produits en cause, Lucentis et Avastin, sont commercialisés sur l’ensemble du territoire de l’Union européenne. Les procédures administratives préalables à la commercialisation de ces médicaments ont d’ailleurs été menées au niveau européen : Avastin et Lucentis ont en effet tous deux fait l’objet d’une AMM européenne, délivrée conformément aux dispositions de la directive n° 2001/83/CE du 6 novembre 2001. De même, les procédures de modification du résumé des caractéristiques produit (« RCP ») de ces deux médicaments ont été mises en œuvre au niveau européen, sous l’égide du comité des médicaments à usage humain (« CHMP »). En outre, la question de l’efficacité et la sécurité comparées de ces deux médicaments dans le traitement de la DMLA a donné lieu à des études scientifiques dans plusieurs pays européens, comme la France (étude GEFAL) et le Royaume-Uni (étude IVAN).
609. En deuxième lieu, les entreprises qui distribuent ces produits, respectivement Novartis et Roche, appartiennent à des groupes pharmaceutiques de dimension internationale, qui sont présents via leurs filiales dans la plupart des États européens.
610. En dernier lieu, les pratiques en cause, qui ont été mises en œuvre au niveau national, ont été susceptibles d’avoir une influence sur les ventes d’Avastin et de Lucentis au-delà du territoire français, et donc d’avoir affecté la structure du marché au niveau européen. En effet, des pratiques de communication, comme celles mises en œuvre au titre du grief n° 1 et du grief n° 2, sont de nature à influencer la structure d’un marché en contribuant à favoriser l’usage de certains produits au détriment de produits concurrents, dans la mesure où ces pratiques sont intervenues dans le contexte d’un débat existant à l’échelle européenne sur l’efficacité et la sécurité comparées des deux médicaments. Dans ces conditions, la décision d’une autorité nationale de santé de favoriser ou de restreindre l’usage d’Avastin dans le traitement de la DMLA, de même que les prises de position de groupes d’études dont l’expertise est reconnue au-delà des frontières nationales, ont pu affecter les ventes des médicaments concernés dans plusieurs pays européens.
611. D’ailleurs, les pratiques en cause s’inscrivaient dans le cadre d’une stratégie européenne, voire mondiale, mise en œuvre par l’entité collective composée par Novartis, Roche et Genentech (cf. paragraphes 713 à 725 de la présente décision). À cet égard, il convient de souligner que l’Autorita Garante della Concorrenza e del Mercato (AGCM) a, dans l’affaire italienne Roche/Novartis, fait application du droit de l’Union, les laboratoires Roche et Novartis ayant été sanctionnés sur le fondement de l’article 101, paragraphe 1, du TFUE, pour avoir mis en place une entente anticoncurrentielle « par objet » en vue de générer une différence artificielle entre les spécialités Avastin et Lucentis pour le traitement de la DMLA (cf. paragraphes 236 à 240 ci-dessus).
612. Il résulte de ce qui précède que les pratiques en cause sont susceptibles d’affecter de manière sensible le commerce entre États membres et d’être qualifiées au regard de l’article 102 du TFUE.
D. SUR LE BIEN-FONDE DES GRIEFS NOTIFIES
1. S’AGISSANT DE LA DELIMITATION DU MARCHE PERTINENT
a) Le marché de produits
Principes applicables
613. Dans sa communication sur la définition du marché en cause aux fins du droit communautaire de la concurrence (JOCE 1997, C 372, p. 5 ; ci-après, la « Communication de la Commission sur la définition du marché en cause »), la Commission européenne a indiqué qu’« un marché de produits en cause comprend tous les produits et/ou services que le consommateur considère comme interchangeables ou substituables en raison de leurs caractéristiques, de leur prix et de l’usage auquel ils sont destinés » (point 7). Suivant la même approche, l’Autorité a rappelé dans son rapport annuel 2011 que « le marché est défini comme le lieu sur lequel se rencontrent l’offre et la demande de produits ou de services spécifiques, considérés par les acheteurs ou les utilisateurs comme substituables entre eux, mais non substituables aux autres biens et services offerts » (page 106).
614. Ainsi que le souligne la Cour de justice dans l’arrêt Generics (UK) rendu sur question préjudicielle, « il ressort de la jurisprudence que la notion de marché pertinent implique qu’une concurrence effective puisse exister entre les produits ou les services qui en font partie, ce qui suppose un degré suffisant d’interchangeabilité en vue du même usage entre tous les produits ou les services faisant partie d’un même marché. L’interchangeabilité ou la substituabilité ne s’apprécie pas au seul regard des caractéristiques objectives des produits et des services en cause. Il convient également de prendre en considération les conditions de la concurrence et la structure de la demande et de l’offre sur le marché (arrêt du 23 janvier 2018, F. Hoffmann-La Roche e.a., C-179/16, EU:C:2018:25, point 51 ainsi que jurisprudence citée) » (arrêt de la Cour de justice du 30 janvier 2020, Generics (UK), aff. C-307/18, point 129).
615. Le secteur des médicaments présente à cet égard une particularité, en ce que la décision d’achat n’est pas prise par l’utilisateur final, mais par le médecin prescripteur, qui choisit le médicament devant être administré à son patient. Dans un arrêt du 15 juin 1999, Lilly France (pourvoi n° 97-15185), la Cour de cassation a approuvé la cour d’appel de Paris qui avait considéré que « l’interchangeabilité des médicaments ne dépend pas fondamentalement de leur identité physique ou chimique, mais de leur interchangeabilité fonctionnelle du point de vue du dispensateur, et donc, dans le cas des médicaments soumis à prescription, également du point de vue des médecins établis ».
616. Afin de déterminer le marché de produits pertinent, et d’apprécier la substituabilité d’un médicament par rapport à un autre, il convient donc de prendre en compte les conditions de la concurrence et la structure de la demande et de l’offre sur le marché, évaluées au regard notamment du point de vue des médecins prescripteurs, celui-ci est très largement dépendant des indications et contre-indications thérapeutiques des médicaments, ou encore des particularités des patients ou du système de santé concerné.
617. Par ailleurs, dans son arrêt rendu sur une question préjudicielle dans l’affaire italienne Roche/Novartis (arrêt du 23 janvier 2018, Hoffmann-Laroche, C-179/16), la Cour de justice s’est prononcée sur la possibilité d’inclure un médicament utilisé « hors AMM » dans le même marché pertinent que celui des médicaments autorisés pour le traitement d’une pathologie donnée. Plus spécifiquement, elle a jugé qu’« une autorité nationale de la concurrence peut inclure dans le marché pertinent, outre les médicaments autorisés pour le traitement des pathologies concernées, un autre médicament dont l’AMM ne couvre pas ce traitement, mais qui est utilisé à cette fin et présente ainsi un rapport concret de substituabilité avec les premiers. Pour déterminer si un tel rapport de substituabilité existe, cette autorité doit, pour autant qu’un examen de la conformité du produit en cause aux dispositions applicables régissant sa fabrication ou sa commercialisation a été effectué par les autorités ou les juridictions compétentes pour ce faire, tenir compte du résultat de cet examen, en évaluant ses éventuels effets sur la structure de la demande et de l’offre » (point 67). Plus spécifiquement, la Cour de justice a souligné que, pour appartenir au même marché de produits, les médicaments ne doivent pas être fabriqués ou vendus de manière illicite (point 52) et que le fait qu’un médicament soit fréquemment prescrit en dehors de son indication révélait l’existence d’un rapport concret de substituabilité avec les spécialités autorisées dans cette indication (point 66). Cette appréciation portait en outre sur les médicaments utilisés pour le traitement de la DMLA, qui sont en cause dans la présente espèce.
Appréciation en l’espèce
618. Les pratiques en cause dans la présente affaire concernent les médicaments Lucentis et Avastin, anti-VEGF utilisés pour le traitement de pathologies oculaires, en particulier pour le traitement de la DMLA exsudative.
619. En effet, en 2007, Lucentis a été autorisé pour le traitement de cette seule pathologie. Ce n’est qu’après 2011 que les indications de Lucentis ont été étendues au traitement d’autres pathologies oculaires (cf. paragraphes 78 et 80 de la présente décision). Avastin a, quant à lui, en dépit de son indication en oncologie, été utilisé « hors AMM » par de nombreux médecins dans le domaine ophtalmique, en particulier pour le traitement de la DMLA (cf. en particulier, paragraphes 65 à 67 de la présente décision).
Le marché du traitement de la DMLA exsudative
620. Dans sa recommandation de juin 2012, la HAS préconise, pour le traitement de la DMLA, le recours aux anti-VEGF. Plus spécifiquement, elle mentionne les anti-VEGF disposant d’une AMM pour le traitement de la DMLA, soit le ranibizumab (Lucentis) et le pegaptanib (Macugen). Par ailleurs, la HAS a présenté deux autres spécialités susceptibles d’être utilisées : le bevacizumab (Avastin), en utilisation « hors AMM », et l’aflibercept (Eylea), alors en cours d’autorisation.
621. Concernant les autres formes de traitement, la HAS indique que « Les contre-indications aux anti-VEGF sont exceptionnelles et les alternatives thérapeutiques doivent être discutées au cas par cas » (cote 14380).
622. La recommandation de la HAS conduit donc à considérer que les anti-VEGF constituent le traitement de première intention de la DMLA exsudative.
623. Ceci est confirmé par les déclarations des ophtalmologistes interrogés au cours de l’instruction. À titre d’illustration, le chef de service en ophtalmologie au CHNO des Quinze-Vingt et à la fondation ophtalmologique Rothschild a déclaré : « A titre liminaire, je tiens à rappeler que la classe thérapeutique des anti-VEGF a révolutionné la prise en charge de la DMLA néovasculaire » (cote 13584, voir également les cotes 13589, 13594 et 13602). Ainsi, à compter de son arrivée sur le marché, en 2007, Lucentis a été – et demeure – le traitement de référence. Par ailleurs, Eylea a été régulièrement utilisé, très rapidement après son entrée sur le marché français, en 2013 (cotes 13584 et 7089, cf. paragraphes 84 à 86 et 183 ci-dessus). Enfin, Avastin était également fréquemment utilisé pour le traitement de la DMLA et d’autres pathologies oculaires, à tout le moins jusqu’à la publication de l’instruction de la DGS de juillet 2012 (cf. paragraphes 65 à 67 et 173 à 180 ci-dessus).
624. Néanmoins, Roche, Genentech et Novartis soutiennent qu’Avastin et les autres anti-VEGF autorisés dans le traitement de la DMLA, tels que Lucentis et Eylea, n’appartiennent pas au même marché de produits. Elles estiment notamment que le fait qu’un nombre très minoritaire de médecins ait choisi de prescrire Avastin en lieu et place de Lucentis ne saurait suffire à établir un rapport concret de substituabilité entre ces deux médicaments, qui présentent des caractéristiques objectives très différentes. Elles affirment en outre que le cadre réglementaire et les prises de position des autorités de santé n’autorisaient les médecins à prescrire Avastin pour le traitement de la DMLA que dans des situations très rares, voire même, prohibaient un tel usage. Roche produit d’ailleurs une étude économique, démontrant, selon lui, que les volumes d’Avastin prescrits après l’introduction de la RTU, venue légitimer l’usage d’Avastin « hors AMM », seraient restés très faibles (autour de 1 % selon les données PMSI(36) exploitées par cette étude) tandis que les parts de marché d’Eylea ont considérablement augmenté dès son entrée sur le marché (pour atteindre près de 40 % selon les données exploitées).
625. Toutefois, en premier lieu, le fait qu’Avastin et Lucentis ne présentent pas les mêmes caractéristiques objectives (ils diffèrent de fait, en termes de classe ATC(37), de propriétés moléculaires, de conditionnement, de même mode d’action, et de mode de délivrance) n’est pas suffisant pour conclure que ces deux spécialités n’appartiennent pas au même marché pertinent. En effet, en dépit du fait que ces deux médicaments n’ont pas été autorisés pour la même indication thérapeutique, ils ont été utilisés, de façon régulière et pendant plusieurs années, par des médecins opérant dans les services les plus en pointe pour le traitement des affections oculaires, pour la même indication : le traitement de la DMLA exsudative.
626. D’abord, plusieurs médecins ont indiqué utiliser Avastin en dehors de l’indication prévue dans son AMM, avant, mais également après, l’arrivée de Lucentis sur le marché. À titre d’illustration, le chef de service de pharmacie clinique du groupe hospitalier Hôpitaux universitaires Paris Centre indiquait, dans un courrier de janvier 2012 adressé à la DGS, que cet établissement avait « fait le choix d’utiliser Avastin (bevacizumab) en dehors de l’AMM plutôt que Lucentis (ranibizumab) selon les modalités de l’AMM, non seulement pour des raisons économiques (Lucentis est en effet 40 fois plus coûteux que notre préparation et cette attitude nous a permis d’économiser 3,5 millions d’euros en 5 ans) mais en fondant notre analyse sur diverses publications scientifiques » (cote 7105). Cet usage, loin d’être une pratique marginale, émanait de praticiens reconnus (les KOL), opérant dans des centres de pointe pour le traitement des affections oculaires (par exemple, les Hospices civils de Lyon, qui est l’un des centres les plus reconnus en ophtalmologie en France).
627. À ce sujet, Novartis reconnaît d’ailleurs dans ses observations en réponse à la notification de griefs que, lorsque Lucentis est arrivé sur le marché en 2007, ce médicament s’est heurté « à une pratique existante » et devait lutter « contre une habitude prise par certains médecins qui utilisaient Avastin » (paragraphe 773). Cette pratique est confirmée par le point d’information de l’AFSSAPS de septembre 2009 dans lequel celle-ci indique, au sujet d’Avastin : « Son mode d’action anti-VEGF, identique à celui des médicaments ayant une AMM en ophtalmologie (Macugen et Lucentis), conduit des ophtalmologistes à utiliser Avastin en injections intravitréennes dans le traitement des atteintes oculaires avec néovascularisation, telles que la dégénérescence maculaire liée à l’âge (DMLA) » (cote 48387) ou encore : « Avastin possède une structure chimique proche de celle de Lucentis, le ranibizumab étant une fraction modifiée de bevacizumab. Cette similarité entre les deux molécules a amené les praticiens à considérer qu’Avastin pouvait être efficace dans les indications ophtalmologiques, en particulier dans le traitement de la DMLA » (cote 48388).
628. Ainsi, la pratique consistant à utiliser Avastin pour le traitement de la DMLA était liée à la conviction, largement partagée par l’AFSSAPS, d’un certain nombre de médecins, en France comme dans d’autres pays (Italie, Grande-Bretagne, etc.) de son efficacité pour cette indication.
629. Ensuite, les estimations internes de Novartis attestent d’une utilisation conséquente d’Avastin dans le traitement de la DMLA. À titre d’illustration, en janvier 2008, Novartis évaluait la part de marché d’Avastin en France à 15 % (cf. paragraphe 180 ci-dessus ; voir également les cotes 2368, 4705, 13977 et 13978 pour d’autres estimations internes). Contrairement à ce que soutient Roche, ces estimations, qui reposent sur des données de sondage d’ophtalmologistes spécialistes de la rétine (i.e. panel Rekam) dont l’échantillon est, certes, de petite taille, conduisent à des intervalles de confiance pour la part de marché d’Avastin relativement larges (de 8 à 22 % en janvier 2008 et de 6 à 20 % en octobre 2008), mais suffisants pour attester d’une utilisation significative d’Avastin dans le traitement de la DMLA(38). Au-delà des estimations de parts de marché, ces documents internes présentent tous Avastin comme concurrent de Lucentis (voir également cotes 2431 et 45134). De plus, aucun document ne conteste la fiabilité de ces données de parts de marché, qui ont, au contraire, été utilisées de façon répétée par Novartis pour la détermination de sa propre stratégie interne, le laboratoire commandant lui-même régulièrement ces sondages auprès d’organismes d’études (cotes 3673 et 4705), ce qui atteste qu’il leur prêtait une valeur probante suffisante.
630. Par ailleurs, les chiffres d’utilisation de l’Hôtel-Dieu, reproduits dans un document interne de Novartis de juin 2010, montrent que le nombre d’injections oculaires réalisées en 2009 par un seul établissement hospitalier (1 029 injections oculaires d’Avastin, dont près de 650 pour le traitement de la DMLA, cote 13995) était nettement plus important que celui extrait de la base de données PMSI pour cette même année (moins de 500 injections ophtalmologiques d’Avastin dont moins de 100 en DMLA dans les établissements publics, cotes 51689 à 51693), les données de l’Hôtel-Dieu étant d’ailleurs déclaratives et probablement sous-estimées selon les termes mêmes dudit document : « Source : Physician declarative data, probably underestimated » (cote 13995).
631. De plus, outre qu’elles sont contradictoires avec les éléments susvisés du dossier, les données PMSI, utilisées par Roche dans son étude économique, présentent de nombreuses limites, qui conduisent à leur retirer toute force probante pour apprécier, dans les circonstances de l’espèce, l’usage d’Avastin, compte tenu des modalités et de l’objet des collectes de données réalisées pour alimenter cette base de données.
632. En effet, les données PMSI, dont le but est de collecter les données sur l’activité hospitalière, retraitées par Roche pour calculer les volumes d’Avastin utilisés pour le traitement de la DMLA, ne couvrent qu’imparfaitement l’utilisation « hors AMM » d’Avastin dans le traitement de la DMLA. D’une part, elles ne prennent pas en compte l’utilisation d’Avastin « hors AMM » dans le cadre du GHS. Or, compte tenu des spécificités du système de tarification à l’activité, une injection intra-vitréenne d’Avastin pouvait être intégrée dans le GHS et ne faire l’objet d’aucun recueil. En ce sens, le chef du service de pharmacie clinique du G.H. Hôpitaux Universitaire de Paris Centre a confirmé que l’usage d’Avastin était pris sur son propre budget sans que celui-ci lui soit remboursé, et donc sans qu’il soit inscrit en sus et codé au PMSI (« l’Hôpital ne gagnait pas plus puisqu’il prenait en charge l’Avastin sur son propre budget sans que celui-ci ne soit remboursé », cote 7096). En outre, la base de données PMSI permet d’assurer un suivi des remboursements par l’Assurance maladie et non d’effectuer un suivi médical des traitements administrés : elle n’a de sens que pour la transmission d’actes codés qui ouvrent droit à remboursement. D’autre part, l’usage d’Avastin « hors AMM » n’était pas réservé aux hôpitaux. La pharmacie à usage intérieur de l’Hôtel-Dieu a, par exemple, développé une pratique consistant à reconditionner Avastin en seringue afin que ces conditionnements soient ensuite utilisés par les ophtalmologues pour des traitements dans des cabinets de ville, comme en atteste un document interne de Novartis de juin 2010 (« internal use + supplier of 50 centers in wAMD and other indications », cote 13995). De même, un courrier électronique interne de Novartis montre que, dans le cadre d’une étude d’impact de la RTU pour l’année 2013, où Novartis examine non seulement les conséquences du PLFSS sur les ventes de Lucentis à l’hôpital, mais également celles aux médecins de ville (« office based »), le laboratoire reconnaît qu’Avastin était utilisé « hors AMM » pour le traitement de la DMLA en ville (cote 50409). Or, les volumes d’Avastin utilisés en ville ne sont pas comptabilisés au PMSI.
633. Enfin, les prises de position des autorités de santé confirment qu’Avastin était fréquemment utilisé « hors AMM » pour le traitement de la DMLA. Par exemple, le protocole d’étude GEFAL rappelle qu’AVASTIN était « largement utilisé de façon non réglementée pour traiter la DMLA dans les établissements publics mais également par les ophtalmologistes du secteur privé. Les nombreuses publications de cas rapportés ou de séries de cas témoignent de l’utilisation du bevacizumab [AVASTIN] hors AMM dans le monde et en France » (soulignement ajouté, cote 2280). De même, dans son courrier électronique du 10 septembre 2008, relançant Roche afin que celui-ci lui communique les échantillons nécessaires à la réalisation des études de stabilité de la solution Avastin reconditionnée en seringue, dans la perspective de l’étude GEFAL, l’AFSSAPS constate : « je pense que ces contrôles sont absolument nécessaires, en raison de l’utilisation importante hors AMM du produit » (soulignement ajouté, cote 16403).
634. Dès lors, contrairement à ce que soutiennent Roche, Genentech et Novartis, il est constant qu’Avastin était fréquemment prescrit pour le traitement de la DMLA et d’autres pathologies oculaires, en dépit du fait que son AMM ne couvrait pas ces indications. En outre, plusieurs documents internes de Novartis attestent du fait que l’usage d’Avastin « hors AMM » pour le traitement de la DMLA était perçu comme une menace potentielle pour Lucentis, que les pratiques en cause visaient à contrer (voir notamment les paragraphes 259 à 260 et 359 de la présente décision). De telles circonstances révèlent par conséquent l’existence d’un rapport concret de substituabilité entre Avastin et les autres anti-VEGF autorisés pour le traitement de la DMLA (et les autres pathologies oculaires concernées), dont Lucentis.
635. En second lieu, contrairement à ce que soutiennent Roche, Genentech et Novartis, l’utilisation d’Avastin « hors AMM » en ophtalmologie, et plus particulièrement pour le traitement de la DMLA, n’était pas illicite.
636. À ce sujet, la Cour de justice a jugé, dans un arrêt rendu sur question préjudicielle concernant l’affaire italienne Roche/Novartis (arrêt du 23 janvier 2018, Hoffmann-Laroche, C-179/16) : « afin d’évaluer dans quelle mesure un produit pharmaceutique dont l’AMM ne couvre pas le traitement de certaines pathologies est substituable à ou interchangeable avec un autre, autorisé quant à lui pour le traitement desdites pathologies, et si lesdits produits relèvent dès lors du même marché pertinent au sens rappelé aux points 50 et 51 du présent arrêt, l’autorité nationale de la concurrence doit, pour autant qu’un examen de la conformité du produit en cause aux dispositions applicables régissant sa fabrication ou sa commercialisation a été effectué par les autorités ou les juridictions compétentes pour ce faire, tenir compte du résultat de cet examen, en évaluant ses éventuels effets sur la structure de la demande et de l’offre » (point 61). Elle en a conclu que, dans la présente affaire, « l’état d’incertitude entourant la licéité des conditions de reconditionnement et de prescription de l’Avastin en vue du traitement de pathologies oculaires ne s’opposait pas à ce que l’AGCM, aux fins de l’application de l’article 101, paragraphe 1, TFUE, conclue que ce produit relevait du même marché qu’un autre médicament dont l’AMM couvre spécifiquement ces indications thérapeutiques » (point 64). Or, cette question préjudicielle avait été transmise par le Conseil d’État italien dans le cadre d’une affaire qui portait précisément sur les mêmes médicaments que dans la présente affaire et sur des pratiques mises en œuvre par les mêmes entreprises (Novartis et Roche) afin de lutter contre le développement, en Italie, de l’usage d’Avastin « hors AMM », de préférence à Lucentis, pour le traitement de la DMLA, pour des raisons de coûts.
637. En l’espèce, le cadre réglementaire français n’interdisait pas l’utilisation d’Avastin « hors AMM » pour le traitement de pathologies oculaires, comme le traitement de la DMLA.
638. En effet, premièrement, contrairement à ce que soutiennent les sociétés mises en cause, l’article L. 5121-1 du code de la santé publique (qui définissait les « préparations hospitalières » comme suit : « tout médicament, à l'exception des produits de thérapies génique ou cellulaire, préparé selon les indications de la pharmacopée et en conformité avec les bonnes pratiques mentionnées à l'article L. 5121-5, en raison de l'absence de spécialité pharmaceutique disponible ou adaptée par une pharmacie à usage intérieur d'un établissement de santé (…) ») n’interdisait pas aux pharmacies à usage intérieur d’hôpitaux de reconditionner Avastin pour un usage en ophtalmologie.
639. Une pharmacie à usage intérieur d’hôpital pouvait ainsi être amenée à préparer des seringues d’Avastin, dès lors que le médecin estimait qu’il n’existait pas de spécialité pharmaceutique adaptée disposant d’une autorisation sur le marché, ce qui a été confirmé par le Conseil d’État dans son arrêt du 24 février 2017 (rendu à l’occasion d’un recours pour excès de pouvoir contre la décision du directeur général de l’ANSM établissant la RTU pour Avastin, CE, 1e et 6e sous-sections réunies, n° 392459, inédit, cf. paragraphe 72 de la présente décision).
640. Ainsi, même en présence d’une spécialité disponible sur le marché, telle que Lucentis, si le médecin estimait que le recours à la spécialité Avastin était le traitement le plus adéquat, le reconditionnement réalisé par une pharmacie intérieure d’hôpital ne contrevenait pas aux dispositions de l’article L. 5121-1 du code de la santé publique.
641. Deuxièmement, l’article L. 5121-12-1 du code de la santé publique, dans sa rédaction issue de la loi Bertrand, n’interdisait pas non plus toute utilisation « hors AMM » d’Avastin pour des indications pour lesquelles Lucentis disposait d’une AMM.
642. En effet, comme cela ressort du paragraphe 69 de la présente décision, si, en pratique, cette évolution législative a réduit les possibilités pour les médecins ophtalmologistes d’utiliser Avastin pour le traitement de la DMLA, dès lors qu’il existait une spécialité disposant d’une AMM pour cette indication, en l’occurrence Lucentis, un médecin pouvait toujours, après la loi Bertrand, considérer que Lucentis n’était pas approprié pour son patient et choisir de lui administrer Avastin, dans l’intérêt de ce dernier. Ceci a également été confirmé par le Conseil d’État dans son arrêt susvisé du 24 février 2017 : « Considérant que, eu égard au développement de la pratique de prescriptions de certaines spécialités en dehors des indications ou des conditions d'utilisation de leur autorisation de mise sur le marché, aux bénéfices susceptibles d'en être attendus ainsi qu'aux risques courus, le législateur a entendu, par l'élaboration de recommandations temporaires d'utilisation, et sans que le deuxième alinéa du I de l'article L. 5121-12-1 du code de la santé publique interdise ces prescriptions en l'absence de recommandation, renforcer les garanties associées à cette pratique par la mise à disposition des médecins, par l'Agence nationale de sécurité du médicament et des produits de santé chargée de leur élaboration, d'informations relatives notamment aux bénéfices attendus de la spécialité et aux risques courus dans l'indication ou les conditions d'utilisation en cause et par la mise en place d'un suivi des patients » (soulignement ajouté). En effet, cette faculté d’administrer aux patients des spécialités en dehors de l’indication au titre de laquelle l’AMM a été délivrée découle du principe supérieur de liberté de prescription, qui a valeur législative et figure parmi les règles déontologiques fondamentales. Le médecin, dans le cadre de cette liberté de prescription, doit être guidé par l’intérêt du patient.
643. Ainsi, après l’entrée en vigueur de la loi Bertrand, un médecin pouvait toujours décider de prescrire Avastin pour un patient atteint de DMLA ou d’une autre pathologie de l’œil, dès lors qu’il jugeait indispensable, compte tenu des données acquises de la science, le recours à cette spécialité.
644. Troisièmement, concernant les prises de position des autorités de santé, la situation a été évolutive. Jusqu’en juillet 2012, les autorités de santé françaises ont adopté une posture de prudence concernant l’utilisation d’Avastin « hors AMM » en ophtalmologie, mais sans l’interdire (voir notamment le point d’information de l’AFSSAPS de septembre 2009 et la recommandation de la HAS de juin 2012, cf. paragraphes 149 à 152 et 160 à 164 de la présente décision). En revanche, en juillet 2012, la DGS a effectivement enjoint aux autorités régionales de santé (ci-après « ARS ») d’interdire aux hôpitaux les préparations de seringues d’Avastin pour une injection intravitréenne, quelles que soient les pathologies soignées (cf. paragraphes 165 à 168 de la présente décision). Le champ de l’instruction de la DGS a toutefois été réduit par une instruction rectificative d’août 2012, précisant qu’Avastin pouvait être utilisé pour les indications pour lesquelles Lucentis ou aucun autre médicament n’avait d’AMM (cf. paragraphes 169 à 171 de la présente décision).
645. Or, si l’instruction de la DGS a conduit la plupart des médecins ophtalmologistes à cesser leur utilisation d’Avastin pour le traitement de pathologies oculaires, cette instruction n’a toutefois pas totalement mis fin à l’usage d’Avastin « hors AMM » en France (cf. paragraphes 178 à 180 de la présente décision).
646. En effet, compte tenu de leur obligation légale de délivrer le traitement le plus adapté à chaque patient, les médecins demeuraient libres, en vertu du principe de liberté de prescription, de décider du médicament à prescrire le plus approprié, compte tenu des données acquises de la science. Roche indique d’ailleurs sur ce point, dans ses observations en réponse au rapport que « le principe de liberté de prescription des médecins, de nature réglementaire, s’impose aux autorités de santé qui, en tant qu’autorités administratives, n’ont pas le pouvoir d’interdire aux médecins la prescription d’un médicament légalement commercialisé sur le marché français, fût-elle pour un usage hors AMM, s’il constitue le traitement le plus adapté ».
647. En outre, l’Autorité ne saurait être liée par une analyse du contexte qui pourrait avoir été retenue par une autre autorité administrative à la suite d’un comportement susceptible d’être anticoncurrentiel (arrêt de la cour d’appel de Paris du 11 juillet 2019, société Janssen-Cilag SAS, n° 18/01945, point 98) et ce, d’autant moins en l’espèce, qu’il ressort des éléments du dossier que les pratiques des sociétés mises en cause ont directement contribué à l’adoption de la circulaire précitée de la DGS (cf. paragraphe 973 de la présente décision).
648. Au surplus, et en tout état de cause, Avastin demeurait, après la publication de l’instruction de la DGS, un concurrent potentiel de Lucentis pour le traitement de la DMLA.
649. En effet, comme le rappellent de façon constante les juridictions européennes, « afin d’apprécier si une entreprise absente du marché se trouve dans un rapport de concurrence potentielle avec une ou plusieurs entreprises déjà présentes sur ce marché, il convient de déterminer s’il existe des possibilités réelles et concrètes que cette première intègre ledit marché et concurrence la ou les secondes » (arrêt de la Cour de justice du 30 janvier 2020, Generics (UK), aff. C-307/18, point 36).
650. Or, dès la fin du mois de juin 2012, les autorités publiques ont commencé à évoquer la possibilité d’une « RTU économique » pour Avastin dans la DMLA (cf. paragraphe 70 de la présente décision) et, jusqu’à l’adoption effective de la RTU pour Avastin au mois de juin 2015, cette controverse n’a jamais cessé et Avastin a continué d’être considéré par Novartis comme une menace pour Lucentis (cf. paragraphe
359 de la présente décision).
651. Ainsi, contrairement à ce que soutiennent Roche, Genentech et Novartis, le contexte réglementaire entourant les conditions de reconditionnement et de prescription d’Avastin pour le traitement de pathologies oculaires, comme le traitement de la DMLA, n’empêche pas de constater que ce médicament relève du même marché que Lucentis ou tout autre médicament dont l’AMM couvre spécifiquement cette indication thérapeutique, dès lors qu’il est constant que, tout au long de la période, Avastin a été utilisé par de nombreux ophtalmologues pour le traitement de la DMLA exsudative et que cette utilisation n’était pas illicite.
652. Par conséquent, il convient de définir un marché du traitement de la DMLA exsudative en première intention, comprenant l’ensemble des anti-VEGF utilisés dans cette indication, incluant Avastin utilisé « hors AMM ».
Les autres marchés du traitement de pathologies oculaires
653. Les anti-VEGF sont utilisés pour le traitement d’autres pathologies oculaires, comme le traitement de l’OMD, l’OBVR ou OVCR, ou encore celui de la baisse visuelle due à une NVC.
654. Comme cela ressort des paragraphes 78 et 86 de la présente décision, Lucentis et Eylea ont obtenu progressivement des AMM pour ces indications. Or, avant que Lucentis obtienne cette AMM, Avastin était largement utilisé pour leur traitement (cote 45084), ce qui est confirmé par plusieurs médecins auditionnés (voir notamment, cote 13585 : « nous l’avons [Avastin] utilisé pour les indications non couvertes par l’AMM de Lucentis », voir également les cotes 7090, 7095, 7096 et 13589).
655. Ainsi, comme pour le traitement de la DMLA, le traitement par anti-VEGF constitue, pour chacune de ces indications, un choix thérapeutique de première intention. Dès lors, Lucentis, Eylea et Avastin doivent être considérés comme entretenant un rapport concret de substituabilité pour le traitement de chacune de ces indications.
656. Par conséquent, il convient de définir autant de marchés qu’il existe d’indications pour le traitement par anti-VEGF.
657. En outre, comme l’indique une note du 2 août 2012 de la société française d’ophtalmologie (« SFO »), Lucentis et Avastin sont utilisés « hors AMM » pour de nombreuses indications rares pour lesquelles il n’existe pas d’autre traitement, comme le « glaucome néovasculaire (rubéose irienne) » ou encore la « rétinopathie des prématurés » (cote 13479). Un tableau fourni par le ministère de la santé donne la liste de ces indications (cote 13515).
658. Conformément à ce qui précède, chacune de ces indications spécifiques pourrait faire l’objet d’un marché pertinent. Toutefois, il n’est pas nécessaire d’opérer une délimitation définitive pour ces indications dans la présente affaire.
La connexité entre les différents marchés
659. En premier lieu, comme cela a été rappelé ci-dessus, les laboratoires commercialisant des anti-VEGF ont obtenu, progressivement, des AMM pour chacune de ces indications autres que la DMLA. De même, l’anti-VEGF Avastin a été largement utilisé « hors AMM » pour le traitement de l’ensemble de ces différentes indications.
660. En second lieu, l’ensemble des pathologies mentionnées ci-dessus concernent des pathologies rétiniennes traitées principalement par injections dans l’œil d’anti-VEGF. Compte tenu de leur mode d’administration identique, l’examen de la question de la tolérance de ces spécialités est indifférent à la pathologie exacte pour laquelle chacune est utilisée.
661. Par conséquent, il convient de relever la connexité existant entre, d’une part, le marché français du traitement de la DMLA exsudative par anti-VEGF et, d’autre part, les autres marchés d’indications traitées par anti-VEGF.
b) La distinction entre le marché de la ville et le marché hospitalier
Principes applicables
662. L’Autorité distingue habituellement le marché de la ville du marché hospitalier. Ainsi, dans sa décision n° 10-D-02 du 14 janvier 2010 relative à des pratiques mises en œuvre dans le secteur des héparines à bas poids moléculaire, elle a considéré qu’une telle distinction était justifiée pour les raisons suivantes : « en ville, les prix sont régulés alors que sur le marché hospitalier les prix sont libres. Par ailleurs, si l’offre est la même, la demande est différente : pour le marché de la ville, la demande intermédiaire est constituée par les grossistes et les pharmacies, et pour le marché de l’hôpital, par les établissements hospitaliers, publics (par exemple, les hôpitaux de l’Assistance publique) ou privés (cliniques privées). Par ailleurs, l’élasticité-prix des acheteurs n’est pas la même : à l’hôpital elle est forte car le prix d’achat affecte le budget des hôpitaux tandis qu’en ville elle est faible, car le patient n’assume pas directement le prix de l’héparine qui lui est remboursé par l’Assurance maladie » (paragraphe 55 ; voir également les décisions n° 13-D-11 du 14 mai 2013 relative à des pratiques mises en œuvre dans le secteur pharmaceutique, point 302 ; n° 13-D-21 du 18 décembre 2013 relative à des pratiques mises en œuvre sur le marché français de la buprénorphine haut dosage commercialisée en ville, point 330 ; et n° 17-D-25 du 20 décembre 2017 relative à des pratiques mises en œuvre dans le secteur des dispositifs transdermiques de fentanyl, point 392).
663. Dans cette même décision, l’Autorité a également indiqué que pour certaines spécialités pharmaceutiques, vendues en ville et à l’hôpital, il pouvait exister « un effet source » des prescriptions à l’hôpital réalisées ensuite en ville (cf. paragraphe 52 de la présente décision) qui permettrait de considérer que les deux marchés sont connexes (décisions n° 10-D-02 du 14 janvier 2010 relative à des pratiques mises en œuvre dans le secteur des héparines à bas poids moléculaire, point 57 et n° 17-D-25 du 20 décembre 2017 relative à des pratiques mises en œuvre dans le secteur des dispositifs transdermiques de fentanyl, point 393).
Appréciation en l’espèce
664. S’il existe des différences importantes entre les conditions de distribution des médicaments en ville et à l’hôpital, identifiées dans la pratique décisionnelle passée (concernant le circuit d’approvisionnement, la structure de la demande et les conditions de négociation du prix), la distinction entre le marché de la ville et le marché de l’hôpital doit être fortement atténuée en l’espèce, compte tenu des particularités des produits concernés.
665. Contrairement à ce que soutiennent Roche, Genentech et Novartis, Avastin et Lucentis entretiennent bien un rapport de concurrence, à l’hôpital comme en ville.
666. En effet, en premier lieu, eu égard aux comportements des médecins hospitaliers (qui demandent aux patients d’acheter Lucentis en ville pour bénéficier du remboursement de la sécurité sociale, y compris lorsque l’injection est réalisée à l’hôpital, cf. paragraphe 176 de la présente décision), les praticiens hospitaliers avaient le choix entre utiliser Avastin dans le cadre d’une hospitalisation, ou prescrire Lucentis dans le cadre d’une consultation externe.
667. En deuxième lieu, si Avastin est un médicament de réserve hospitalière, plusieurs pièces du dossier attestent du fait que ce médicament pouvait néanmoins être administré en ville (cf. paragraphe 632 ci-dessus).
668. En troisième lieu, les documents internes de Novartis présentent constamment Avastin comme un concurrent de Lucentis, soulignant en particulier la menace que représentait Avastin sur le prix de Lucentis (cf. paragraphes 259 à 260, 291 et 359 ci-dessus). En effet, comme cela ressort des paragraphes 34 à 42 ci-dessus, le Comité économique des produits de santé (« CEPS ») pouvait s’appuyer sur la concurrence d’Avastin pour négocier à la baisse le prix de Lucentis.
669. Roche soutient que l’utilisation « hors AMM » d’Avastin ne pouvait exercer une pression concurrentielle sur le prix de Lucentis. Toutefois, plusieurs éléments au dossier montrent que le CEPS aurait pu faire pression pour obtenir une baisse du prix de Lucentis et ce, quand bien même Avastin n’avait pas été clairement admis comme un comparateur à Lucentis. En particulier, un document interne de Novartis atteste du fait que des députés sont intervenus auprès du CEPS, en mars 2014, pour l’inciter fortement à réévaluer la situation tarifaire de Lucentis sur la question de la comparaison Lucentis / Avastin (« some French Parliament representatives put pressure on Mah [market autorisation holder] to reassess the situation in general including RTU/Avastin », cote 45130), ce qui est confirmé par les débats parlementaires sur la LFSS de 2013. Le président du CEPS précisait en outre, dans un échange de courriels, qu’en cas de désaccord, il pouvait fixer le prix par une décision administrative (cote 45124).
670. L’étude économique présentée par Roche, portant sur l’élasticité-prix de la demande de Lucentis en ville et à l’hôpital entre 2008 et 2018, et selon laquelle la demande des anti-VEGF serait effectivement inélastique au prix en ville et significativement élastique à l’hôpital, ne saurait remettre en cause ce constat.
671. En effet, s’agissant des médicaments remboursés à 100 %, les patients, les pharmaciens et les grossistes en ville sont peu sensibles aux prix dès lors que, s’agissant des patients, ils ne supportent pas le coût associé à la prise de médicaments, ceux-ci étant remboursés par la sécurité sociale. S’agissant des grossistes et des pharmaciens, leurs marges sont administrées et assises sur le prix fabricant hors taxes PFHT, le prix net des remises confidentielles accordées par les industriels à l’Assurance maladie n’intervenant pas dans la détermination des marges des distributeurs. À l’inverse, les acheteurs hospitaliers sont, quant à eux, sensibles aux prix des médicaments, qui impactent directement leur budget.
672. Mais cette sensibilité différenciée des deux types de demandeurs aux prix ne remet pas en cause, en l’espèce, la conclusion relative à l’existence d’une relation de concurrence entre Avastin et Lucentis, aussi bien en ville, au travers, essentiellement, des négociations entre le CEPS et Novartis autour du prix de Lucentis (cf. paragraphes 30 à 43 ci-dessus), qu’à l’hôpital, puisque Lucentis n’était jamais acheté par les hôpitaux mais fournis par des pharmacies de ville.
673. En outre, pour les raisons évoquées précédemment, les comportements mis en œuvre par les membres de l’entité collective composée de Novartis, Roche et Genentech étaient de nature à affecter tant le segment de l’hôpital que celui de la ville. En effet, quand bien même Avastin est un médicament de réserve hospitalière, il faut relever que ce médicament a été utilisé en ville, d’une part, compte tenu des différences de coûts des traitements Lucentis et Avastin, et d’autre part, parce que des praticiens estimaient l’administration d’Avastin plus adaptée à l’état de santé de leurs patients. En outre, le CEPS pouvait s’appuyer sur l’équivalence thérapeutique d’Avastin pour négocier à la baisse le prix de Lucentis. La préservation du niveau de prix de Lucentis était à cet égard une des motivations principales des pratiques de Novartis s’agissant de l’usage « hors AMM » d’Avastin (cf. paragraphes
259 à 260, 291 et 359 ci-dessus).
674. Au regard de ce qui précède, il convient de considérer que, compte tenu des particularités, notamment de prescription et d’achat, des produits concernés, il convient, en l’espèce, de ne pas distinguer le marché de la ville et le marché de l’hôpital.
675. Au demeurant, même si l’Autorité devait distinguer les marchés de la ville et de l’hôpital en l’espèce, les abus mis en œuvre par les membres de l’entité collective seraient, en tout état de cause, de nature à affecter ces deux segments ou marchés connexes, la délimitation d’un seul ou de deux marchés étant également sans incidence sur la détermination de la position dominante collective détenue par Novartis, Roche et Genentech.
c) Le marché géographique
Principes applicables
676. Dans sa communication précitée, la Commission européenne a précisé que « Le marché géographique en cause comprend le territoire sur lequel les entreprises concernées sont engagées dans l’offre des biens et des services en cause, sur lequel les conditions de concurrence sont suffisamment homogènes et qui peut être distingué de zones géographiques voisines parce que, en particulier, les conditions de concurrence y diffèrent de manière appréciable » (point 8).
677. Il ressort de la pratique décisionnelle constante des autorités de concurrence en matière de marchés pharmaceutiques que les marchés de médicaments sont généralement de dimension nationale (voir, notamment, les décisions n° 10-D-02 du 14 janvier 2010 relative à des pratiques mises en œuvre dans le secteur des héparines à bas poids moléculaire, point 52 ; n° 13-D-11 du 14 mai 2013 relative à des pratiques mises en œuvre dans le secteur pharmaceutique, point 308 ; n° 13-D-21 du 18 décembre 2013 relative à des pratiques mises en œuvre sur le marché français de la buprénorphine haut dosage commercialisée en ville, point 333, ainsi que la décision de la Commission européenne du 4 février 1998, 98/526/CE, Hoffman-LaRoche/Boehringer Mannheim). Dans la décision n° 10-D-02 du 14 janvier 2010 relative à des pratiques mises en œuvre dans le secteur des héparines à bas poids moléculaire, l’Autorité a ainsi constaté que « le marché géographique pertinent d’une spécialité, vendue aux hôpitaux mais également en ville, et dont le prix de vente en pharmacie est régulé par les pouvoirs publics, est celui du territoire national ».
Appréciation en l’espèce
678. Lucentis et Eylea sont remboursés par le système de sécurité sociale français et se sont vus attribuer par le CEPS un prix ouvrant droit à remboursement par l’assurance maladie. Ils sont vendus sur l’ensemble du territoire national.
679. Concernant Avastin, comme indiqué précédemment, le niveau de pression concurrentielle que ce produit pouvait faire peser sur les autres anti-VEGF était étroitement lié à l’organisation des systèmes nationaux de sécurité sociale et de remboursement des médicaments, ainsi qu’au cadre juridique de l’usage « hors AMM » des médicaments.
680. Il convient donc de considérer que le marché du traitement de la DMLA exsudative par anti-VEGF et les autres marchés d’indications traitées par anti-VEGF sont de dimension nationale.
d) Conclusion concernant le marché pertinent
681. Il ressort de ce qui précède que les marchés concernés sont le marché français du traitement de la DMLA exsudative par anti-VEGF et les marchés français des autres indications traitées par anti-VEGF, combinant la ville et l’hôpital, compte tenu des particularités de l’espèce.
682. Au demeurant, même si l’Autorité devait distinguer les marchés de la ville et de l’hôpital en l’espèce, elle constaterait, en tout état de cause, que les abus mis en œuvre par les membres de l’entité collective étaient de nature à affecter ces deux segments ou marchés connexes, la délimitation d’un seul ou de deux marchés étant également sans incidence sur la détermination de la position dominante collective détenue par Novartis, Roche et Genentech.
2. S’AGISSANT DE LA DOMINANCE COLLECTIVE DE ROCHE, NOVARTIS ET GENENTECH
a) Principes applicables
683. L’existence d’une position dominante collective est établie lorsque « les entreprises en cause ont, ensemble, notamment en raison de facteurs de corrélation existant entre elles, le pouvoir d’adopter une même ligne d’action sur le marché et d’agir dans une mesure appréciable indépendamment des autres concurrents, de leur clientèle et, finalement, des consommateurs » (voir notamment, les décisions n° 06-D-18 du 28 juin 2006 relative à des pratiques mises en œuvre dans le secteur de la publicité cinématographique et n° 12-D-06 du 26 janvier 2012 relative à des pratiques mises en œuvre dans le secteur des agrégats et des marchés aval à Saint-Pierre et Miquelon ; et les arrêts de la Cour de justice du 31 mars 1998, aff. jointes C-68/94 et C-30/95, Kali & Saltz, point 221 et du Tribunal du 25 mars 1999, aff. T-102/96, Gencor, point 163).
684. Pour déterminer si Roche, Novartis et Genentech disposent d’une position dominante collective sur le marché du traitement de la DMLA exsudative, il faut examiner, en premier lieu, si elles constituent une entité collective puis, en second lieu, examiner si cette entité détient une position dominante sur le marché en cause.
Sur l’existence d’une entité collective
685. Pour déterminer si plusieurs entreprises peuvent être considérées comme une entité collective, il convient d’apporter la preuve qu’elles ont des liens juridiques ou économiques.
686. En effet, dans l’arrêt Compagnie maritime belge, la Cour de justice a indiqué qu’une position dominante est susceptible d’être détenue « par plusieurs entités économiques, juridiquement indépendantes l’une de l’autre, à condition que, du point de vue économique, elles se présentent ou agissent ensemble sur un marché spécifique, comme une entité collective » (arrêt de la Cour de justice du 16 mars 2000, Cie maritime belge, aff. C-395/96 et 396/96, point 36). Ainsi, pour constituer une entité collective, les entreprises doivent être « suffisamment liées entre elles pour adopter une même ligne d’action » (arrêts de la Cour de justice du 27 avril 1994, Almelo, C-393/92, point 42 ; du 15 octobre 1995, DIP, C-140/94 et a., point 26 ; du 5 octobre 1995, Centro Servizi Spediporto, C-96/94, point 33). Le Tribunal a indiqué en ce sens, dans l’affaire Atlantic Container Line, que « tel est le cas si ces entreprises sont en mesure de prévoir leurs comportements réciproques et sont donc fortement incitées à aligner leur comportement sur le marché de façon, notamment, à maximiser leur profit commun (…) » (arrêt du Tribunal du 30 septembre 2003, Atlantic Container Line, Aff. T-191/98 et a., point 595).
687. Les liens ou facteurs de corrélation pris en considération peuvent être soit juridiques, soit économiques. Dans l’affaire Compagnie maritime belge, la Cour de justice a ainsi précisé que l’existence d’une position dominante collective peut résulter « de la nature et des termes d’un accord, de la manière de sa mise en œuvre et, partant des liens ou facteurs de corrélation entre entreprises qui en résultent. Toutefois, l’existence d’un accord ou d’autres liens juridiques n’est pas indispensable (…) [une telle constatation] pourrait résulter d’autres facteurs de corrélation et dépendrait d’une appréciation économique, et notamment, d’une appréciation de la structure du marché » (arrêt de la Cour de justice du 16 mars 2000, Cie maritime belge, aff. C-395/96 et 396/96, point 45).
688. De même, dans une décision n° 06-D-02 du 20 février 2006 relative à des pratiques relevées dans le secteur des travaux routiers liées à la fabrication d’enrobées bitumeux dans le département des Ardennes, le Conseil de la concurrence a indiqué : « L’existence de liens structurels entre des entreprises d’une part, tels que des liens en capital ou encore des accords formalisés entre elles, et l’adoption d’une ligne commune d’action sur le marché d’autre part, suffisent à démontrer l’existence d’une position de dominance collective (CJCE, 16 mars 2000, Compagnie maritime belge ; TPI, 7 octobre 1999, Irish Sugar ; Cour de cassation, 5 mars 1996, Total Réunion Comores ; cour d’appel de Paris, 30 octobre 2001, OMVESA ; cour d’appel de Paris, 4 juin 2002, CFDT Radio Télé). En l’absence de tels liens, la seule structure du marché peut permettre de mettre en évidence une position dominante collective, si les critères cumulatifs dégagés par le Tribunal de première instance dans son arrêt Airtours du 6 juin 2002 (affaire T-342/99) sont réunis, à savoir la structure oligopolistique et la transparence du marché concerné, la possibilité d’exercer des représailles sur les entreprises déviant de la ligne d’action commune et enfin la non-contestabilité du marché ou l'absence de compétition potentielle » (points 107 à 109 ; voir également les décisions n° 06-D-18 du 28 juin 2006 relative à des pratiques mises en œuvre dans le secteur de la publicité cinématographique, point 180 et n° 12-D-06 du 26 janvier 2012 relative à des pratiques mises en œuvre dans le secteur des agrégats et des marchés aval à Saint-Pierre et Miquelon, point 193).
Sur l’existence d’une position dominante
689. Comme en matière d’abus de position dominante individuelle, il est nécessaire de déterminer si l’entité collective a le pouvoir, compte tenu de sa position particulière sur le marché, de s’abstraire du comportement de ses concurrents et clients.
690. Dans sa décision n° 10-D-36 du 17 décembre 2010 relative à des pratiques mises en œuvre dans le secteur du gaz de pétrole liquéfié (GPL) conditionné, l’Autorité a indiqué que : « Le concept de dominance collective renvoie donc à un groupe d’entreprises qui disposent, en commun, d’un pouvoir de marché assimilable à celui d’une entreprise en position dominante simple, c’est-à-dire lui permettant de s’abstraire de la concurrence d’autres entreprises actives sur le même marché. Le concept de dominance collective réunit ainsi deux notions : en premier lieu celle de l’interdépendance, et en second lieu celle, plus classique, de la dominance. (…) A cet égard, les juridictions européennes ont rappelé à plusieurs reprises que l’examen d’une dominance collective doit notamment s’apprécier au regard de la capacité du groupe d’entreprises considéré de se comporter sur le marché en cause, dans une mesure appréciable, de façon ‘‘indépendante’’ vis-à-vis de leurs concurrents, de leurs clients et des consommateurs (voir, notamment, l’arrêt de la Cour de justice des Communautés européennes du 16 mars 2000, Compagnie maritime belge transports et autres/Commission, affaires jointes C-395/96 P et C-396/96 P, Rec. p. I-1365, points 33 et 34) » (points 36 et 38).
691. Selon une jurisprudence constante, l’appréciation de la position dominante d’une entreprise s’effectue à partir d’un faisceau de critères qui prend en compte des données d’ordre structurel, comme les parts de marché de l’entreprise et celles de ses principaux concurrents, mais aussi des éléments qui sont de nature à donner un avantage concurrentiel à l’entreprise concernée, comme l’appartenance à un groupe puissant ou la détention d’une avance technologique.
692. La cour d’appel de Paris a ainsi relevé que « l’existence d’une position dominante résulte en général de la réunion de facteurs divers, qui, pris isolément, ne seraient pas nécessairement déterminants mais qui traduisent ensemble la puissance économique requise par la jurisprudence » (arrêt de la cour d’appel de Paris du 18 décembre 2014, Sanofi, n° 2013/12370, page 7). L’Autorité a rappelé dans sa décision n° 13-D-11 du 14 mai 2013 relative à des pratiques mises en œuvre dans le secteur pharmaceutique qu’« une telle position peut résulter de différents facteurs caractérisant le marché lui-même ou l’entreprise, comme la détention, soit d’un monopole légal ou de fait sur une activité, soit de parts de marché substantielles. Une telle position peut aussi résulter de l’appartenance à un groupe de grande envergure, de la faiblesse des concurrents, de la détention d’une avance technologique ou d’un savoir-faire spécifique » (point 311).
693. S’agissant du pouvoir de marché, en dehors de toute situation de monopole, l’Autorité a souligné, dans sa décision n° 13-D-11, précitée, que, « D’une manière générale, l’examen des parts de marché constitue un paramètre essentiel dans l’appréciation de la dominance éventuelle d’une entreprise sur son marché. Il ressort de la jurisprudence des juridictions communautaires et nationales que des parts de marché extrêmement importantes constituent par elles-mêmes, et sauf circonstances exceptionnelles, la preuve de l’existence d’une position dominante » (point 314). La Cour de justice considère en effet que la possession, dans la durée, d’une part de marché extrêmement importante constitue, sauf circonstances exceptionnelles, la preuve de l’existence d’une position dominante, et que tel est le cas d’une part de marché de plus de 50 % (arrêts du 13 février 1979, Hoffmann-La Roche/Commission, C-85/76, point 41, du 3 juillet 1991, Akzo/Commission, C-62/86, Rec. p. I-3359, point 60, et du 6 décembre 2012, AstraZeneca e.a./Commission, C-457/10 P, point 176).
694. Cependant, comme l’a souligné l’Autorité à plusieurs reprises, l’analyse de la position dominante d’une entreprise ne saurait toujours se limiter à un examen des parts de marché. D’autres facteurs peuvent être pris en compte, en particulier lorsque l’importance de la part de marché détenue par l’entreprise ne suffit pas à elle seule à caractériser sa position dominante sur le marché. Il pourra ainsi être tenu compte du rapport entre les parts de marché détenues par l’entreprise concernée et celles détenues par ses concurrents, de l’intensité de la concurrence et des barrières à l’entrée sur le marché concerné ou encore des caractéristiques propres à l’entreprise en cause : leadership (ou capacité d’influence) sur le marché, image de marque, puissance financière (voir, à cet égard, les décisions n° 10-D-32 du 16 novembre 2010 relative à des pratiques dans le secteur de la télévision payante, paragraphe 532, n° 10-D-02 du 14 janvier 2010 relative à des pratiques mises en œuvre dans le secteur des héparines à bas poids moléculaire, et n° 13-D-11 du 14 mai 2013 relative à des pratiques mises en œuvre dans le secteur pharmaceutique, paragraphe 316).
695. En ce qui concerne les autres facteurs, la cour d’appel de Paris a relevé, dans une affaire concernant le secteur pharmaceutique, d’une part, que « le brevet initial que cette société [Sanofi Aventis] a exploité en monopole pendant dix ans et l’importance du groupe dans lequel elle s’inscrit, lui ont permis d’acquérir une réputation de référence, accentuée en l’espèce par le retour d’expérience de dix ans qu’elle peut faire, et fait, valoir auprès des professionnels de santé », et d’autre part, que « les visites aux médecins permises par l’important réseau de visiteurs médicaux dont disposait la société Sanofi Aventis, lors de la mise en œuvre des pratiques reprochées, constituait un incontestable outil d’influence auprès des prescripteurs » (arrêt de la cour d’appel de Paris du 18 décembre 2014, Sanofi, n° 2013/12370, page 7).
b) Appréciation en l’espèce
696. Il convient d’examiner successivement si Genentech, Roche et Novartis constituent une entité collective, au sens défini ci-dessus, puis la position détenue par celle-ci sur le marché du traitement de la DMLA exsudative.
Sur l’existence d’une entité collective réunissant Genentech, Roche et Novartis
697. Conformément aux principes exposés ci-dessus, il convient d’examiner, d’abord, les liens structurels existants entre Genentech, Roche et Novartis, pour ensuite, déterminer si ces entreprises ont pu adopter une ligne commune d’action.
Les liens structurels entre Genentech, Roche et Novartis
698. Il ressort des éléments du dossier que Genentech, Roche et Novartis sont liées par un tissu serré de liens structurels importants et stratégiques (cf. paragraphe 220 de la présente décision).
699. D’une part, Genentech est lié à Roche et Novartis par le biais de contrats de licence portant sur la commercialisation, respectivement, d’Avastin et de Lucentis,
700. En effet, comme indiqué précédemment, le laboratoire Genentech a été à l’origine de la recherche sur les anti-VEGF, et développé, dans ce cadre, le ranibizumab (Lucentis) et le bevacizumab (Avastin). Le laboratoire, qui assure la commercialisation de ces deux spécialités sur le territoire américain, a accordé des licences pour chacun de ces produits, dans le reste du monde, respectivement à Novartis et à Roche.
701. Les contrats liant Genentech et Novartis, d’une part, et Roche, d’autre part, prévoient un système très organisé de remontées d’informations, de forums de discussions et de comités communs de gestion.
702. En effet, comme souligné aux paragraphes 199 à 209 de la présente décision, si l’accord de licence liant Genentech et Roche pour Avastin confère à ce dernier une certaine autonomie, notamment s’agissant des relations avec les autorités d’enregistrement en dehors des États-Unis et des efforts de promotion et de commercialisation du médicament, celle-ci intervient dans un cadre très contraint. En particulier, la commercialisation d’Avastin ne peut être effectuée que dans le cadre d’un plan de développement défini très en amont, comprenant les indications initiales, ainsi que les autres indications pouvant être développées (cote 50401). Dans ce cadre, Roche est tenu d’informer Genentech, régulièrement, sur la politique commerciale et le positionnement d’Avastin sur le territoire concédé (cote 50400). Ce dispositif de remontée d’informations est en outre complété par les nombreux contacts intervenant entre les deux laboratoires, dans le cadre des différents comités (comité de commercialisation conjointe, comité de gestion, comité de développement, comité financier conjoint) créés par l’accord cadre les liant (cotes 45041 et 45042).
703. De même, s’agissant de l’accord de licence conclu entre Genentech et Novartis pour Lucentis, comme souligné aux paragraphes 210 à 220 de la présente décision, si chaque entreprise est responsable des activités réglementaires et de commercialisation de Lucentis sur son territoire respectif, cette autonomie s’inscrit dans un cadre très structuré, dans lequel les deux laboratoires s’accordent sur les principales décisions concernant la vie du produit, qu’il s’agisse du développement de nouvelles indications, de la constitution du dossier scientifique ou du positionnement commercial du produit. En particulier, si les deux laboratoires définissent ensemble un plan de développement, correspondant au traitement des maladies oculaires (cotes 15512 et 45138), Novartis ne peut organiser ou soutenir des études en dehors de ce champ (cote 15530). L’accord prévoit, par ailleurs, un grand nombre d’obligations d’information et de consultation entre Novartis et Genentech, sur tous les aspects de la vie de Lucentis (cotes 15495 et 15496, cotes 45114 et 45115), ainsi que de nombreux contacts dans le cadre de comités (comité de gestion conjointe et équipe de projet conjoint) institués par l’accord (cotes 45139 et 15560).
704. Roche, Genentech et Novartis soutiennent sur ce point que les échanges d’informations organisés dans le cadre desdits contrats ne sauraient être retenus pour caractériser une position dominante collective, dans la mesure où ils sont limités à ce qui est nécessaire entre un donneur et un preneur de licence, et n’excèdent pas le cadre traditionnel d’un contrat de licence standard. Toutefois, s’il est exact qu’un échange d’informations entre un preneur et un donneur de licence sur le développement et la commercialisation d’un produit donné en licence relève, en principe, de la mise en œuvre classique des relations commerciales entre les sociétés concernées, il n’en demeure pas moins que l’existence de telles obligations est susceptible de figurer parmi les indices retenus pour caractériser l’existence d’une entité collective.
705. D’autre part, Genentech, Roche et Novartis ont des liens capitalistiques croisés particulièrement significatifs.
706. En effet, comme souligné aux paragraphes 188 et 220 de la présente décision, Roche était l’actionnaire majoritaire de Genentech jusqu’en 2009 et détient depuis cette date la totalité de son capital. Par ailleurs, comme souligné au paragraphe 195 de la présente décision, Novartis détient, pour sa part, une participation non contrôlante dans le capital de Roche, avec 6,2 % du capital (les actions sans droit de vote représentant 81,5 % du capital de Roche Holding) et 33,33 % des droits de vote de Roche Holding (cote 47658).
La ligne d’action commune de Genentech, Roche et Novartis
707. La structure contractuelle et capitalistique existant entre les laboratoires Genentech, Roche et Novartis leur a permis d’adopter une ligne d’action commune sur le marché, concernant la commercialisation de Lucentis et d’Avastin.
708. En premier lieu, les liens structurels présentés ci-dessus ont permis à Genentech, Roche et Novartis de connaître leurs comportements respectifs, et, de ce fait, de s’assurer qu’ils adoptaient la même ligne d'action.
709. En effet, les dispositions contractuelles susvisées – et plus particulièrement, les comités communs de gestion et les échanges d’informations qu’elles organisent – assurent une véritable transparence entre le donneur de licence et ses licenciés sur leurs politiques commerciales, juridiques et administratives respectives. Plus spécifiquement, Roche et Novartis ont la possibilité de connaître les éléments essentiels de leurs politiques commerciales respectives, ainsi que l’état de leurs relations avec les autorités de santé, compte tenu du rôle central de Genentech (révélé notamment par son implication dans les pratiques sanctionnées au titre du grief n° 2, cf. paragraphes 1018 à 1028 de la présente décision). En outre, le contrôle exercé par Roche sur Genentech lui permet, à tout le moins, une transparence de l’information avec sa filiale.
710. Novartis fait valoir que les contrats conclus avec Genentech organisent une certaine autonomie des parties sur leurs territoires respectifs, s’agissant des activités commerciales et réglementaires. Toutefois, il n’en demeure pas moins que ces contrats organisent une collaboration poussée sur tous les aspects de la vie des produits concernés (qu’il s’agisse du développement de nouvelles indications, du lancement de nouvelles études, des relations avec les autorités publiques, des communications scientifiques ou encore du positionnement commercial de chaque produit). En particulier, comme le souligne d’ailleurs Novartis dans ses observations, le laboratoire ne dispose en pratique d’aucune autonomie pour décider de développer de nouvelles indications pour Lucentis, si cela devait entrer en contradiction avec le plan de développement défini en commun avec Genentech.
711. Ainsi, quand bien même chacun des deux licenciés a la possibilité théorique de prévoir des développements autonomes s’agissant de la molécule pour laquelle il est titulaire d’une licence, l’économie des contrats signés prévoit que les parties impliquées doivent suivre un plan de développement commun. Il est donc impossible que l’une des entreprises prépare le développement d’un médicament dans une nouvelle indication, ou modifie son discours commercial et scientifique, sans que les autres en soient informées.
712. Enfin, si Roche et Novartis n’ont pas de relation directe au titre de ces différents contrats de licence, le fait que Genentech se trouve au coeur de ce système contractuel lui a permis de faire la synthèse des enjeux économiques, juridiques et scientifiques pour les deux molécules, et d’établir une politique commune, ce que démontrent notamment les échanges entre la « Avastin team » et la « Lucentis team » au moment de l’annonce par le gouvernement français de ses projets de réforme législative sur la question des RTU (cf. paragraphes 20 à 28 et 474 de la présente décision). Ainsi, Genentech, qui formulait pour le monde entier les principes de positionnement respectif d’Avastin et de Lucentis, est directement intervenue dans la détermination des réponses à apporter au débat public concernant la commercialisation de ses produits, Avastin et Lucentis, en France. Dans ce contexte, Roche et Novartis ont développé un discours très similaire, fondé sur les mêmes éléments techniques, fournis en grande partie par Genentech (cf. paragraphes 1018 à 1028 de la présente décision).
713. En second lieu, il existait une forte incitation financière pour les trois laboratoires à ne pas s'écarter de la ligne de conduite commune sur le marché, visant à limiter les prescriptions « hors AMM » d’Avastin au profit des ventes de Lucentis.
714. En effet, compte tenu des différences de prix entre les deux spécialités et de la pratique consistant à fabriquer plusieurs seringues avec un seul flacon d’Avastin (cf. paragraphe 66 de la présente décision), toute utilisation d’Avastin à la place de Lucentis pour une injection dans l’œil était susceptible d’entraîner un manque à gagner significatif pour chacun des trois laboratoires concernés. D’abord, pour Novartis, qui reçoit, en tant que licencié, le produit des ventes de Lucentis sur le marché concerné : son intérêt commercial réside dans la maximisation des ventes de ce produit. Ensuite, pour Genentech, qui perçoit, en tant que donneur de licence, les redevances des ventes de Lucentis sur le marché concerné : à doses équivalentes, ce laboratoire perçoit moins de revenus en licence pour les ventes d’Avastin en ophtalmologie que pour celles de Lucentis. Enfin, pour Roche, qui, en tant qu’actionnaire principal puis unique depuis mars 2009 de Genentech, profite des bénéfices de sa filiale : ses intérêts sont donc alignés sur ceux du laboratoire américain.
715. Ainsi, compte tenu de la détention capitalistique de Genentech par Roche, les politiques commerciale et de développement de ces deux entités sont très étroitement liées, Genentech dépendant de Roche pour la fixation des orientations générales de son action. Dans ce contexte, Roche n’avait aucun intérêt à poursuivre une politique consistant à positionner Avastin sur le marché d’une façon qui ne correspondait pas aux intérêts de sa filiale Genentech.
716. Roche et Genentech soutiennent néanmoins que le fait que Genentech ne soit pas présent sur le marché français du traitement de la DMLA exsudative et que Roche serait quasi-absent du marché ferait échec à ce constat. De même, Novartis conteste, d’une part, l’intérêt commercial de Genentech à maintenir une distinction entre les indications d’Avastin et de Lucentis et, d’autre part, que cet intérêt aurait été partagé par Novartis et Roche.
717. Toutefois, il ressort de la jurisprudence qu’une position dominante collective est susceptible d’être caractérisée alors même que les entreprises concernées ne sont pas actives sur les mêmes marchés (arrêts de la Cour de justice du 27 avril 1994, Almelo, C-393/92 et du Tribunal du 7 octobre 1999, Irish Sugar, T-228/97).
718. Or, en l’espèce, si Genentech n’est pas actif en France, il a développé à la fois le bevacizumab (Avastin) et le ranibizumab (Lucentis) et les exploite tous deux aux États-Unis (cf. paragraphes 61, 63 et 76 de la présente décision). Ainsi, outre la perception des redevances dues par Novartis sur les ventes de Lucentis dans le reste du monde, Genentech avait un intérêt commercial manifeste à favoriser une distinction stricte entre l’usage d’Avastin en oncologie et celui de Lucentis en ophtalmologie, s’agissant des ventes réalisées sur le territoire américain (où Lucentis coûtait 2 000 dollars la dose, tandis que le coût du traitement par Avastin variait entre 50 et 150 dollars(39)). Dans ce contexte, le développement de l’usage d’Avastin pour le traitement de la DMLA, et plus généralement en ophtalmologie, ainsi que la reconnaissance de l’efficacité et la sécurité de cette spécialité par les autorités de santé françaises, était susceptible d’avoir un effet sur le marché américain, en favorisant le recours à ce traitement alternatif à Lucentis. Le débat scientifique sur l’utilisation d’Avastin en ophtalmologie étant mondial (avec la réalisation d’études aux États-Unis, en Grande-Bretagne ou en France notamment, comme en témoignent les études scientifiques présentées aux paragraphes 94 à 128 de la présente décision), Genentech n’avait donc aucune incitation à favoriser la concurrence entre les deux produits sur le marché français.
719. S’agissant de la prétendue quasi-absence de Roche sur le marché du traitement de la DMLA exsudative, il ressort des développements qui précèdent sur la relation de concurrence entre Lucentis et Avastin (cf. paragraphes 618 à 682 de la présente décision) que cet argument manque en fait.
720. Dès lors, le laboratoire Genentech, quoique présent uniquement sur le marché américain, avait bien intérêt à adopter une ligne d’action commune avec ses licenciés, Novartis et Roche, notamment par le biais d’échanges d’informations, de façon à maximiser son profit.
721. Cette ligne d’action commune ressort d’ailleurs de plusieurs pièces du dossier, comme le courrier électronique du 8 mai 2013 adressé par le responsable du produit Lucentis de Genentech à des salariés du groupe Roche, évoquant une rencontre avec Novartis, en vue d’adopter une stratégie de communication commune au moment de la publication de l’étude GEFAL (cf. paragraphe 430 de la présente décision) ou bien les échanges de courriels, fin juin 2012, entre Roche et le « LifeCycle Leader Avastin » de Genentech, présentant la position à tenir sur l’usage « hors AMM » d’Avastin dans le traitement de la DMLA (cf. paragraphes 471 à 475 de la présente décision). Ces éléments démontrent sans équivoque que Genentech intervenait dans la formulation des réponses à apporter au débat public concernant la commercialisation de ses produits, Avastin et Lucentis, en France.
722. Contrairement à ce que soutient Roche, le fait que les dispositions contractuelles des contrats de licence conclus par Genentech avec Roche et Novartis imposent à chacun des licenciés de maximiser le profit de chacun des produits concernés n’est pas de nature à remettre en cause ce constat. D’ailleurs, les éléments démontrant l’implication de Genentech dans l’abus visé au titre du grief n° 2 attestent du fait que c’est le laboratoire américain qui a fourni, en grande partie, les éléments utiles à Roche et à Novartis pour développer et diffuser leur discours insistant sur les risques liés à l’utilisation d’Avastin « hors AMM » en ophtalmologie (voir, les paragraphes 1018 à 1028 de la présente décision).
723. En conséquence, les intérêts financiers de Genentech, Roche et Novartis étaient parfaitement alignés autour de la commercialisation privilégiée de Lucentis, au détriment des ventes d’Avastin.
724. À cet égard, il convient de souligner que, contrairement à ce que soutiennent Roche et Genentech, le fait que les trois laboratoires ne se soient jamais présentés comme une seule et même entreprise ne fait nullement obstacle à ce constat. En effet, il ressort de la jurisprudence susvisée que plusieurs entreprises peuvent être considérées comme constituant une entité collective, même si elles ne se présentent pas formellement comme un seul opérateur, dès lors qu’elles « agissent ensemble comme une entité collective » (cf. paragraphe 686 de la présente décision).
725. Il ressort de ce qui précède que le réseau de contrats de licence mis en place par Genentech, de même que la combinaison de leurs dispositions, des modalités de leur application effective, de l’intérêt de Genentech à maintenir une distinction entre Avastin et Lucentis, ainsi que des liens capitalistiques existant entre Roche et Genentech, ont incité ces entreprises à adopter un comportement coordonné sur le marché, et limité très fortement leurs possibilités d’actions divergentes.
Conclusion sur l’entité collective constituée par Genentech, Roche et Novartis
726. Au regard de ce qui précède, il y a lieu de considérer que Genentech, Roche et Novartis forment une entité collective, pour les besoins de la commercialisation d’Avastin et de Lucentis.
Sur la position dominante détenue par l’entité collective
727. Conformément aux principes exposés ci-dessus, il convient d’examiner si l’entité collective constituée par Genentech, Roche et Novartis disposait d’un pouvoir de marché lui permettant de s’abstraire de la concurrence des autres entreprises présentes sur le marché et de la réaction des clients et consommateurs.
728. Il importe à cet égard d’examiner les parts de marché de l’entité collective, ainsi que les particularités du secteur concerné.
Sur les parts de marché détenues par l’entité collective
729. Les éléments présents au dossier montrent que l’entité collective, formée par Genentech, Roche et Novartis, disposait, à l’époque des pratiques, de parts de marché très importantes sur le marché français du traitement de la DMLA exsudative.
730. En effet, entre 2008 et 2013, parmi tous les médicaments commercialisés pour le traitement de la DMLA exsudative en France (Lucentis, Avastin, Visudyne etMacugen, cf. paragraphes 62 à 91 de la présente décision), seul Macugen – distribuée par Pfizer et ne représentant, selon les estimations internes de Novartis, que 4 % des parts de marché en 2008 (cote 2424) – n’était pas détenu par cette entité.
731. En revanche, après son entrée sur le marché en novembre 2013, Eylea – commercialisé en Europe par le laboratoire Bayer – a capté 36 % des parts de marché en trois mois (cf. paragraphe 183 de la présente décision).
732. Les sociétés mises en cause affirment toutefois, d’une part, que l’entité collective a été contrainte, dans sa ligne de conduite, bien avant l’entrée effective d’Eylea sur le marché en novembre 2013, par la concurrence potentielle que représentait l’arrivée prochaine de ce médicament sur le marché. Plus spécifiquement, Roche, Genentech et Novartis soutiennent qu’Eylea pouvait être considéré comme un concurrent potentiel, exerçant de ce fait une pression concurrentielle sur l’entité collective, dès la fin de l’année 2007, lorsque les essais de phase III pour l’aflibercept (Eylea) ont été lancés.
733. En l’espèce, s’il est exact que plusieurs documents internes de Novartis, datant de 2010 et 2012, mentionnaient la concurrence à venir d’Eylea, force est de constater qu’aucun des éléments du dossier n’atteste de la capacité de Bayer d’accéder au marché avec ce médicament dans un délai à même de faire peser une pression concurrentielle sur l’entité collective au moment des pratiques en cause. Au contraire, le laboratoire commercialisant Lucentis n’anticipait pas une arrivée de ce concurrent avant la fin de l’année 2011, comme en atteste un document interne de Novartis de juillet 2008 (« VEGF Trap Eye (…) Bayer (…) Launch date : Q3-Q4 2011(EU) », cote 2368 ; et « First real competitor : VEGF Trap (Regeneron) will be launched in 2011 », cote 2446). Or, ce médicament ne sera finalement distribué en France qu’à compter de novembre 2013 (cf. paragraphe 86 de la présente décision).
734. Or, conformément à la jurisprudence européenne, « pour qu’un opérateur puisse être qualifié de concurrent potentiel, son entrée potentielle doit pouvoir se faire suffisamment rapidement aux fins de peser et ainsi d’exercer une pression concurrentielle sur les participants au marché » (arrêt de la Cour de justice du 30 janvier 2020, Generics (UK), aff. C-307/18, points 43 et 44). S’agissant plus spécifiquement de l’appréciation de la situation de concurrence potentielle aux fins de l’article 102 du TFUE, la Cour de justice a précisé, s’agissant de la détermination du marché pertinent, qu’un fabricant peut être considéré comme concurrent potentiel s’il est « en mesure de se présenter à brève échéance sur le marché concerné avec une force suffisante pour constituer un contrepoids sérieux » (arrêt de la Cour de justice du 30 janvier 2020, Generics (UK), aff. C-307/18, point 133).
735. Dès lors, même si le développement d’Eylea était connu depuis 2007, ce médicament ne peut être considéré comme ayant constitué, avant novembre 2013, un contrepoids sérieux à l’entité collective composée par Genentech, Roche et Novartis. D’ailleurs, aucun élément du dossier ne permet de constater que l’entrée potentielle sur le marché d’Eylea aurait été de nature à contraindre le comportement et à remettre en cause le plan d’action commun de l’entité collective, dans la mesure où seule sa présence sur le marché comme alternative réelle et immédiate aurait pu permettre à la demande d’éventuellement exercer une contrainte sur l’entité collective.
736. D’autre part, Roche et Genentech soutiennent que seule une position dominante simple de Novartis pourrait, en théorie, être constatée, compte tenu des parts de marché extrêmement élevées de ce laboratoire, de la présence quasi-inexistante de Roche et de l’absence totale de Genentech sur le marché concerné. Roche et Genentech affirment en particulier que c’est uniquement dans le cas où aucune entreprise ne dispose, sur le marché, d’un pouvoir suffisant pour caractériser une position dominante simple qu’il peut être nécessaire de vérifier l’existence d’une position dominante collective.
737. Contrairement à ce que soutiennent les mises en cause, lorsqu’elle se prononce sur un abus de position dominante collective, l’Autorité n’est pas tenue d’examiner préalablement si les opérateurs sur le marché détiennent une position dominante simple.
738. En l’espèce, s’il est vrai que Novartis disposait de parts de marché individuelles très élevées, au cas d’espèce, le laboratoire n’avait pas la possibilité de développer, sur le marché français du traitement de la DMLA exsudative, un comportement indépendant de celui de Roche et Genentech. En effet, la situation « quasi-monopolistique » dont bénéficiait Lucentis dépendait directement de l’attitude de Roche et de son donneur de licence Genentech, concernant l’utilisation d’Avastin en ophtalmologie. Et, ainsi qu’il a été rappelé aux paragraphes 707 à 725 de la présente décision, la stratégie de développement et de gestion du cycle de vie de Lucentis était déterminée en étroite collaboration avec Genentech et Roche à travers, notamment, les comités de gestion.
739. En outre, non seulement le laboratoire Roche ne s’est pas opposé au discours véhiculé par Novartis auprès des professionnels et des autorités de santé, mais il a adopté, à rebours de toute rationalité économique individuelle, une communication similaire à celle de son concurrent, en faveur de l’usage de Lucentis et, de fait, au détriment des ventes de son propre produit, Avastin (cf. paragraphes 843, 894 et 1034 de la présente décision).
740. Par conséquent, contrairement à ce que soutiennent Roche et Genentech, Novartis n’avait pas, en tout état de cause, la capacité de développer un comportement indépendant sur le marché français du traitement de la DMLA exsudative. À l’inverse, comme cela ressort des développements ci-dessus, l’entité collective formée par Genentech, Roche et Novartis n’était confrontée, jusqu’à la fin de l’année 2013, à aucun concurrent susceptible de remettre en cause son pouvoir de marché.
Sur les particularités du secteur et des acteurs concernés
741. En premier lieu, Novartis est, selon ses propres analyses, un laboratoire de référence dans le domaine de l’ophtalmologie sur le marché français (voir notamment les cotes 2424 et 2521), qui a développé des relations particulières avec les différents acteurs du secteur.
742. En effet, Novartis dispose d’un important réseau de visiteurs médicaux et délégués hospitaliers spécialisés, qui entretiennent des relations quotidiennes avec les médecins ophtalmologistes (cf. paragraphe 258 de la présente décision). Il a également mis en place des équipes médicales internes, les MSL (ou « référent médical »), qui lui permettent de détenir une expertise scientifique dans le domaine de l’ophtalmologie et de se positionner comme un interlocuteur crédible vis-à-vis des praticiens (cf. paragraphe 233 de la présente décision). Par ailleurs, il entretient des liens importants avec les médecins les plus en pointe dans le secteur, les KOL (ou « Key opinion leader »), et avec les grands services hospitaliers, en organisant des comités consultatifs, en participant à des congrès et séminaires, et en finançant de nombreux protocoles de recherche (cf. paragraphes 234 et 235 de la présente décision). Enfin, Novartis est un partenaire privilégié des principales associations de patients atteints de DMLA (Retina France et Association DMLA).
743. En deuxième lieu, le secteur du médicament est, compte tenu de l’objectif de protection de santé publique, très réglementé, et se caractérise par des barrières à l’entrée particulièrement élevées.
744. De fait, le processus pour obtenir une AMM ou une extension d’indication pour un médicament est particulièrement long et complexe. Il est en effet nécessaire de conduire, outre le développement de la molécule, des études cliniques de grande ampleur, et de suivre des procédures administratives complexes (octroi de l’autorisation de mise sur le marché (AMM), évaluation du service médical rendu (SMR) et de l’amélioration du service médical rendu (ASMR), fixation du prix, cf. paragraphes 7 à 49 ci-dessus). Il est donc matériellement impossible pour un laboratoire pharmaceutique de conduire ces démarches sans être détecté par le marché. En outre, toute entrée d’un produit concurrent exige au minimum plusieurs années de développement, d’études cliniques et de procédures administratives. Une entrée soudaine d’un produit concurrent, qui pourrait notamment être attiré par des prix élevés concédés au titulaire de l’AMM, est, de ce fait, exclue. À cet égard, comme exposé ci-dessus, une période de six années s’est écoulée entre le commencement des essais de phase III pour l’aflibercept (Eylea) et sa première commercialisation en France.
745. En troisième lieu, l’organisation du système de santé français, et notamment le fait que l’obtention auprès de la HAS d’un niveau d’ASMR élevé est cruciale pour les laboratoires, en ce qu’elle détermine le prix du médicament (cf. paragraphes 30 à 43 de la présente décision, cote 15185) limite fortement les incitations pour un laboratoire à développer un produit pour une indication dans laquelle des produits performants sont déjà commercialisés.
746. En quatrième lieu, concernant les clients des laboratoires pharmaceutiques, et plus particulièrement des médecins, prescripteurs, ceux-ci s’appuient avant tout sur leur diagnostic et leur pratique thérapeutique, afin de déterminer le traitement le plus approprié. Les considérations liées au prix du médicament interviennent ainsi de façon secondaire, même s’il existe certains mécanismes visant à inciter les praticiens à prescrire des médicaments moins chers ou génériques, afin de limiter les dépenses d’assurance maladie, ou si certains médecins peuvent orienter leur choix de prescription en fonction de considérations économiques (cf. paragraphe 66 de la présente décision, cotes 7105 et 7089).
747. En dernier lieu, s’agissant des capacités de réaction de la part des autorités de santé françaises, l’éclatement des compétences entre la HAS et le CEPS notamment, en matière d’évaluation du SMR, d’une part, et d’analyse pharmaco-économique, d’autre part, limite leur possibilité d’une réplique rapide et efficace à l’encontre de pratiques de laboratoires pharmaceutiques telles que celles en cause en l’espèce (cf. paragraphes 30 à 43 de la présente décision).
748. Novartis conteste toutefois ces deux derniers points.
749. S’agissant tout d’abord de la possibilité de réaction des médecins, Novartis affirme, outre le fait que les médecins interrogés ont déclaré ne pas avoir été influencés par le discours du laboratoire – ce qui relève de l’analyse des effets des pratiques notifiées au titre du grief n° 1 et sera donc examiné infra (cf. paragraphes 837 à 888 de la présente décision) –, que les médecins ophtalmologistes pouvaient tenir compte des considérations budgétaires, et ainsi privilégier un traitement moins onéreux, en lien avec leur diagnostic et leur pratique thérapeutique.
750. Il est exact que les motivations des médecins ayant participé à l’étude GEFAL ont pu relever, au moins pour partie, de considérations liées à la prise en compte des contraintes pesant sur les finances publiques (compte tenu du coût extrêmement élevé de Lucentis et du nombre de patients affectés par la DMLA). Toutefois, comme l’a indiqué en audition le Professeur Y…, directeur de l’étude, leur démarche résultait précisément du constat qu’il n’était pas possible de choisir d’utiliser Avastin « hors AMM », en se fondant uniquement sur des considérations financières : « Du côté des médecins, nous ne souhaitions pas dépenser excessivement l’argent public, mais il était évident que c'est normal d'utiliser le produit avec une AMM et ce n’est pas nous qui décidions du prix. A l'époque, il y avait un monopole de Lucentis. C’était la seule molécule qui marchait » (cote 16622).
751. C’est donc parce que les médecins jugeaient nécessaire de trouver une solution pour remédier à cette situation, qu’ils ont choisi de participer à cette étude.
752. S’agissant ensuite de la possibilité de réaction des autorités de santé, Novartis fait valoir que celles-ci disposaient des meilleures compétences scientifiques et disposaient de leur propre capacité de décision.
753. Au demeurant, les pratiques en cause ont été mises en œuvre dans un secteur particulier, caractérisé par une grande aversion au risque, au regard de ses enjeux humains et de santé publique (cf. paragraphes 761 à 768 de la présente décision).
754. Ainsi, des pratiques, telles que celles notifiées, de communication dénigrante et/ou d’immixtion indue dans les processus décisionnels de ces autorités, sont susceptibles d’influer sur les choix des professionnels de santé (médecins et pharmaciens) et des représentants des autorités de santé, quelles que soient par ailleurs les compétences techniques de ces derniers.
755. D’ailleurs, si l’étude GEFAL, et la RTU qui l’a suivie, peuvent être considérées comme une réaction des autorités de santé françaises et d’une partie du corps médical à la situation générée par le prix très élevé de Lucentis, c’est précisément cette réaction que l’entité collective a cherché à retarder par ses pratiques.
756. Par conséquent, compte tenu de ce contexte législatif et réglementaire spécifique et du débat scientifique dans lequel les pratiques se sont inscrites (cf. paragraphes 92 à 171 de la présente décision), avant la publication des méta-analyses reprenant l’ensemble des résultats des études de comparaison, les autorités de santé n’avaient pas la possibilité de mettre en cause le positionnement adopté par l’entité collective.
757. Il résulte de ce qui précède que les caractéristiques du secteur du médicament limitent fortement les possibilités d’entrée rapide d’un concurrent pour une indication donnée et restreignent les capacités des prescripteurs et des autorités de santé à réagir à une politique de prix élevés ou de restriction de l’innovation mise en œuvre par un ou plusieurs laboratoires pharmaceutiques.
758. En conséquence, les caractéristiques du secteur et des acteurs concernés renforçaient les incitations de l’entité collective constituée par Genentech, Roche et Novartis, à suivre une ligne commune d’action.
Conclusion
759. Il ressort de ce qui précède que Genentech, Roche et Novartis détenaient, jusqu’à l’arrivée d’Eylea sur le marché concerné, en novembre 2013, une position dominante collective sur le marché du traitement de la DMLA exsudative.
3. S’AGISSANT DE LA QUALIFICATION DES PRATIQUES
760. Après des remarques préliminaires sur le contexte dans lequel les pratiques ont été mises en œuvre (a), l’Autorité examinera ci-après la pratique de communication de Novartis auprès des professionnels de santé, des associations de patients, du grand public et des autorités de santé (b), puis la pratique de communication de Roche, Novartis et Genentech auprès des autorités de santé (c).
a) Remarques préliminaires sur le contexte des pratiques
761. Avant d’examiner les pratiques reprochées à Genentech, Roche et Novartis, il convient de rappeler le contexte dans lequel celles-ci ont été mises en œuvre, et notamment la prudence, voire l’aversion au risque des professionnels de la santé et des représentants des autorités de santé, qui interviennent dans un secteur tout à fait particulier au regard de ses enjeux humains – la santé des patients et, dans les situations les plus graves, leur survie même dépendent de l’efficacité et de l’innocuité des médicaments qui leur sont prescrits et délivrés – mais également économiques et financiers.
762. Ainsi que l’a souligné la cour d’appel de Paris, « il existe une aversion au risque des professionnels de santé, et notamment des personnes chargées d’élaborer les décisions de l’AFSSAPS, en raison notamment d’une certaine judiciarisation des questions de santé. S’agissant, en particulier de l’AFSSAPS, l’importance de ses décisions sur la santé publique est considérable, et se traduit par une responsabilité non moins importante. Ceci a pour conséquence que toute contestation, fondée ou non, conduit quasi-inéluctablement à un ralentissement du processus décisionnel au sein de l’AFSSAPS, ce qu’aucun laboratoire pharmaceutique ne peut ignorer » (arrêt de la cour d’appel de Paris du 11 juillet 2019, Janssen-Cilag SAS, n° 18/01945, point 273 ; voir également la décision n° 17-D-25 du 20 décembre 2017 relative à des pratiques mises en œuvre dans le secteur des dispositifs transdermiques de fentanyl, points 414 à 419).
763. Dans ce même arrêt, la cour d’appel a précisé que la tenue d’un discours dénigrant par une entreprise en position dominante à l’encontre d’un produit concurrent nouveau est susceptible d’entraver son accès au marché, « a fortiori (…) dans le secteur des médicaments, lorsque le discours dénigrant met en cause l’efficacité ou la sécurité d’un médicament nouveau, compte tenu de l’aversion marquée des professionnels de santé pour le risque, dans un contexte de judiciarisation croissante des questions de santé » (arrêt de la cour d’appel de Paris du 11 juillet 2019, Janssen-Cilag SAS, n° 18/01945, point 411).
764. L’Autorité a fait ce constat à plusieurs reprises (décisions n° 13-D-11, précitée, point 358 ; n° 13-D-21, précitée, point 355 ; et n° 17-D-25, précitée, point 419). Il convient donc de tenir compte, dans l’analyse du discours commercial d’un laboratoire pharmaceutique, « de la forte rigidité au changement des médecins prescripteurs et des pharmaciens, ainsi que de l’aversion aux risques des professionnels de santé » (arrêt de la cour d’appel de Paris du 18 décembre 2014, n° 2013/12370, page 14, confirmé par un arrêt de la Cour de cassation du 18 octobre 2016, n° 15-10384).
765. Par ailleurs, l’Autorité a dans le passé constaté que les médecins ne disposent pas toujours des connaissances précises et complètes sur tous les médicaments disponibles (décisions n° 13-D-11 du 14 mai 2013 relative à des pratiques mises en œuvre dans le secteur pharmaceutique, point 347 ; n° 13-D-21 du 18 décembre 2013 relative à des pratiques mises en œuvre sur le marché français de la buprénorphine haut dosage commercialisée en ville, point 350 ; et n° 17-D-25, précitée, point 415).
766. L’Autorité s’est déjà également prononcée sur le rôle central de la « visite médicale », par les représentants des laboratoires (appelés « visiteurs médicaux »), qui présentent les médicaments aux médecins. Celle-ci constitue en effet, pour les médecins, une source majeure d’information sur les médicaments, en raison de son accessibilité, de sa gratuité et de son interactivité (décisions n° 13-D-11, précitée, points 352 à 357 ; n° 13-D-21, précitée, point 353 ; et n° 17-D-25, précitée, point 417).
767. L’importance de la visite médicale, en tant que source d’information, impose donc à ceux qui l’effectuent, en l’occurrence les visiteurs médicaux, de partager les informations dont ils disposent de façon absolument objective, complète et fiable (décisions n° 13-D-11, précitée, point 357 ; n° 13-D-21, précitée, point 354).
768. Enfin, l’Autorité relève que la mission des agences de santé, qui sont chargées de garantir, au travers de leurs missions de sécurité et de vigilance sanitaire, l'efficacité, la qualité et le bon usage de tous les produits de santé, repose en particulier sur la transmission d’informations par les professionnels de la santé mais aussi par les laboratoires pharmaceutiques (décision n° 17-D-25, précitée, point 420).
b) Concernant la pratique visée par le grief n° 1
Principes applicables
769. Dans un arrêt du 18 décembre 2014, la cour d’appel de Paris a considéré « que les dispositions de droit interne et communautaire prohibant la mise en œuvre de pratiques d’abus de position dominante sont rédigées en termes généraux et que toute pratique, y compris le dénigrement des concurrents actuels ou potentiels, est susceptible de constituer un abus prévu par ces textes dès lors qu’elle a pour objet ou peut avoir pour effet de fausser le jeu de la concurrence (article L. 420-2 du code de commerce) ou qu’elle est susceptible d’affecter le commerce entre États membres de l’Union européenne (article 102 du TFUE) » (arrêt n° 2013/12370, confirmé par l’arrêt de la Cour de cassation du 18 octobre 2016, n° 15-10384).
770. Selon une pratique décisionnelle constante de l’Autorité, le dénigrement peut consister à jeter publiquement le discrédit sur une personne, un produit ou un service identifié. Il se distingue de la critique dans la mesure où il émane d’un acteur économique qui cherche à bénéficier d’un avantage concurrentiel en pénalisant son compétiteur (décisions n° 13-D-11 du 14 mai 2013 relative à des pratiques mises en œuvre dans le secteur pharmaceutique, point 365 ; n° 13-D-21 du 18 décembre 2013 relative à des pratiques mises en œuvre sur le marché français de la buprénorphine haut dosage commercialisée en ville, point 360 et décision n° 17-D-25 du 20 décembre 2017 relative à des pratiques mises en œuvre dans le secteur des dispositifs transdermiques de fentanyl, point 530).
771. Pour qu’un dénigrement puisse être qualifié d’abus de position dominante, il convient que soit établi un lien entre la domination de l’entreprise et la pratique de dénigrement (décision n° 09-D-14 du 25 mars 2009 relative à des pratiques mises en œuvre dans le secteur de la fourniture de l’électricité, points 57 et 58, confirmée par arrêt de la cour d’appel de Paris du 23 mars 2010, Gaz et électricité de Grenoble, n° 2009/09599 ; voir également la décision n° 10-D-32 du 16 novembre 2010 relative à des pratiques dans le secteur de la télévision payante, point 305 et les décisions n° 13-D-11, précitée, point 366 ; n° 13-D-21, précitée, point 361 et n° 17-D-25, précitée, point 531).
772. Pour apprécier l’existence d’une pratique de dénigrement, l’Autorité s’attache, en premier lieu , à vérifier si le discours commercial tenu par l’entreprise en position dominante relève de constatations objectives ou s’il procède d’assertions non vérifiées (décision n° 07-D-33 du 15 octobre 2007 relative à des pratiques mises en œuvre par la société France Télécom dans le secteur de l’accès à Internet à haut débit, point 305 ; voir également les décisions n° 13-D-11, précitée, point 367 ; n° 13-D-21, précitée, point 362 et n° 17-D-25, précitée, point 532).
773. Dans le secteur pharmaceutique, l’Autorité considère que, s’il est parfaitement loisible à un laboratoire de mettre en exergue les qualités objectives d’un produit, le fait de mettre en évidence non pas seulement des qualités, mais des différences qui, dans le contexte du discours tenu et des conditions dans lesquelles il est entendu, ne peuvent se comprendre que comme des différences substantielles, de nature à soulever un doute objectif sur les qualités des spécialités génériques concurrentes ou sur les risques associés à la substitution, peut témoigner d’une volonté d’induire le praticien en erreur et être constitutif d’un abus de position dominante (décisions n° 13-D-11, précitée, point 373 ; n° 13-D-21, précitée, point 365 et n° 17-D-25, précitée, point 533).
774. Cette analyse a été confirmée par la cour d’appel de Paris. Elle a, ainsi, souligné que « d’une part, que la divulgation d’une information de nature à jeter le discrédit sur un concurrent constitue un dénigrement, peu important qu’elle soit exacte (Com., 24 septembre 2013, pourvoi n° 12-19.790), d’autre part, que la divulgation d’une information de nature à jeter le discrédit sur un produit est constitutive de dénigrement, à moins que l’information en cause ne se rapporte à un sujet d’intérêt général et repose sur une base factuelle suffisante, et sous réserve qu’elle soit exprimée avec une certaine mesure (Com., 9 janvier 2019, pourvoi n° 17-18.350) » (arrêt du 11 juillet 2019, Janssen-Cilag SAS, n° 18/01945, point 353 ; voir également arrêt de la Cour de cassation du 4 mars 2020, A.St.A World-Wide, n° 18-15.651). De même, elle a jugé que « la diffusion d’une information négative, voire l’instillation d’un doute sur les qualités intrinsèques d’un médicament, peut suffire à le discréditer immédiatement auprès des professionnels de santé ». En effet, « instiller un doute dans l’esprit de professionnels de la santé, ciblé sur la qualité ou les propriétés du médicament générique, en délivrant des informations incomplètes, ambigües ou présentées de telle manière qu’elles suggèrent l’existence d’un risque à le substituer ou entretiennent, pour des motifs injustifiés, une crainte ou une prévention à cet égard, constitue bien une pratique déloyale et abusive, qui, si elle a pour objet et peut avoir pour effet de restreindre la diffusion sur le marché de ce médicament générique, contrevient aux dispositions (…) du droit de la concurrence » (arrêt de la cour d’appel de Paris du 18 décembre 2014, n° 2013/12370, page 14, confirmé par un arrêt de la Cour de cassation du 18 octobre 2016, n° 15-10384).
775. En lien direct avec le cas d’espèce, la Cour de justice a jugé, dans l’arrêt rendu sur question préjudicielle dans l’affaire Roche/Novartis (arrêt du 23 janvier 2018, Hoffmann-Laroche, C-179/16), que peuvent être qualifiées de trompeuses les informations qui « visaient, d’une part, à induire l’EMA et la Commission en erreur et à obtenir l’ajout de la mention d’effets indésirables dans le résumé des caractéristiques de ce produit, afin de permettre au titulaire de l’AMM d’engager une campagne de communication auprès des professionnels de la santé, des patients et des autres personnes concernées dans le but d’amplifier artificiellement cette perception, et, d’autre part, à exagérer, dans un contexte d’incertitude scientifique, la perception par le public des risques liés à l’utilisation hors AMM de l’Avastin, compte tenu, notamment, du fait que l’EMA et la Commission n’ont pas modifié le résumé des caractéristiques de ce médicament en ce qui concerne ses ‘‘effets indésirables’’, mais se sont bornées à émettre des ‘‘mises en garde spéciales et précautions d’emploi’’ » (point 92).
776. Dans ce sens, la cour d’appel de Paris a souligné « d’une part, que la divulgation d’une information de nature à jeter le discrédit sur un concurrent constitue un dénigrement, peu important qu’elle soit exacte, d’autre part, que ce n’est pas en l’espèce l’objectivité ou l’exactitude des informations contenues dans le discours de la société requérante qui est en cause, mais le lien établi entre deux informations exactes mais indépendantes et présentées de façon incomplète permettant de susciter un doute, voire une crainte sur l’efficacité ou la sécurité des médicaments génériques » (arrêt de la cour d’appel de Paris du 18 décembre 2014, n° 2013/12370, page 15, confirmé par un arrêt de la Cour de cassation du 18 octobre 2016, n° 15-10384).
777. En deuxième lieu, afin de déterminer si le discours commercial de l’entreprise est de nature à influencer la structure de marché, l’Autorité examine les effets attendus ou réels de ces discours auprès des partenaires commerciaux ou de la clientèle potentielle de ses concurrents (décision n° 10-D-32, précitée, point 307 ; voir également les décisions n° 13-D-11, précitée, point 368 ; n° 13-D-21, précitée, point 363 et n° 17-D-25, précitée, point 537). Il n’est en effet pas nécessaire de démontrer que le comportement de l’entreprise en position dominante a eu un effet anticoncurrentiel concret sur le marché concerné pour pouvoir le qualifier d’abusif. Il suffit de démontrer qu’il tend à restreindre la concurrence ou, en d’autres termes, qu’il est de nature à avoir un tel effet (arrêt de la Cour de justice du 17 février 2011, TeliaSonera, aff. C-52/09, point 64 ; voir également l’arrêt de la Cour, AstraZeneca c/ Commission, C-457/10 P, point 112 et la décision n° 13-D-21, précitée, point 363).
778. Or, la cour d’appel de Paris a jugé que « l’expérience enseigne que la tenue d’un discours dénigrant par une entreprise en position dominante à l’encontre d’un produit concurrent nouveau est potentiellement apte à entraver son accès au marché » (arrêt du 11 juillet 2019, Janssen-Cilag SAS, n° 18/01945, point 410).
779. Enfin, au nombre des éléments que l’Autorité retient pour établir l’existence d’un lien entre la domination de l’entreprise en cause et la pratique de dénigrement, figurent la notoriété de cette entreprise et la confiance que lui accordent les acteurs du marché qui sont de nature à renforcer significativement l’impact du discours développé par celle-ci. L’Autorité a ainsi pris en considération le fait que l’entreprise mise en cause bénéficiait, aux yeux du grand public, de la réputation et de la notoriété d’un ancien monopole gérant un service public (décision n° 07-D-33, précitée, point 79 ; voir également les décisions n° 13-D-11, précitée, point 369 ; n° 13-D-21, précitée, point 364 et n° 17-D-25, précitée, point 538).
Appréciation en l’espèce
780. Conformément aux principes exposés ci-dessus, il convient d’examiner si la communication mise en œuvre par Novartis auprès des professionnels de santé, des associations de patients, du grand public et des autorités de santé revêt un caractère trompeur et si ce discours a été de nature à influencer la structure du marché.
781. Plus précisément, il ressort des pièces du dossier que Novartis a diffusé, par le biais d’une campagne de communication globale et structurée, un discours dénigrant, en exagérant, de manière injustifiée, les risques liés à l’utilisation d’Avastin « hors AMM » pour le traitement de la DMLA, et plus généralement en ophtalmologie, en comparaison avec la sécurité et la tolérance de Lucentis pour un même usage.
Sur le caractère trompeur de la communication de Novartis
782. Novartis a mis en œuvre une campagne de communication globale et structurée tendant à jeter le discrédit sur l’utilisation d’Avastin en ophtalmologie, en faveur de Lucentis.
♦ Une communication globale auprès des différents acteurs du secteur de la santé
783. Novartis soutient que le dossier ne contient pas de preuve de la diffusion du discours reproché, ni de ce que ce discours aurait été reçu par les professionnels de santé, les associations de patients, le grand public et les autorités de santé. Le laboratoire souligne qu’au contraire, d’une part, les déclarations des professionnels de santé auditionnés par les services d’instruction montreraient que ceux-ci n’ont pas été destinataires de discours dénigrant de la part de Novartis et, d’autre part, le dossier ne contiendrait aucune audition d’associations de patients susceptible de venir confirmer la diffusion de ce discours à leur endroit. Novartis prétend également que la communication envisagée dans ses documents internes n’était pas proactive, mais destinée à être utilisée uniquement en réaction à des sollicitations extérieures.
784. Toutefois, contrairement à ce que soutient Novartis, il ressort des pièces du dossier que Novartis a effectivement mis en œuvre sa politique de communication active sur le terrain.
785. En effet, en premier lieu, de nombreuses pièces figurant au dossier contiennent un message qui avait vocation à être diffusé.
786. À titre d’illustration, le courriel du 10 mars 2008, adressé par le directeur du marketing en ophtalmologie du groupe Novartis aux salariés en charge du produit Lucentis, auquel est annexé un document portant sur Avastin et Lucentis, précise que : « Le contenu de ce document peut être utilisé par le personnel médical et scientifique de Novartis, ainsi que par les KOL et tout autre personne, afin de défendre les différences entre les deux produits et contre l’argument selon lequel les produits seraient ‘‘essentiellement les mêmes’’ », cote 3630). De même, les « messages clés » et « réponses à objections », figurant dans la présentation de la réunion « Advocacy Lucentis » du 26 mai 2010, étaient destinés à être utilisés par les forces commerciales de Novartis dans le cadre de plans d’actions développés en région (cotes 2137, 2140 et 2141). Par ailleurs, le document, intitulé « Elevator Speech » daté de juin 2010, reprenant les principaux messages clés tendant à insister sur les risques liés à l’utilisation d’Avastin en ophtalmologie, était destiné à être distribué à l’ensemble des forces commerciales de Novartis (cotes 13988 et 13992 ; voir également cote 14850). Tel était également le cas du document, intitulé « Messages clés Lucentis/Avastin » mis à jour en mars 2013 (cote 2982), devant être distribué notamment aux visiteurs médicaux, dont le rôle est précisément de rendre visite aux professionnels de santé.
787. Ainsi, s’il est exact que ces documents indiquent, pour la plupart, qu’ils sont à usage interne et n’ont pas vocation à être diffusés ou remis à l’extérieur, il n’en demeure pas moins que cet avertissement ne concernait que les supports écrits, mais que les messages pouvaient, et même devaient, être diffusés oralement aux professionnels de santé.
788. Plus particulièrement, comme cela ressort de la présentation du 10 avril 2013, le laboratoire a adopté une stratégie de communication différenciée entre, d’une part, les KOL, c'est-à-dire les médecins les plus reconnus dans le domaine du traitement de la DMLA et d’autres maladies oculaires et, d’autre part, les autres médecins pour lesquels la communication était envisagée plutôt de façon réactive (cote 45146).
789. À cet égard, plusieurs pièces figurant au dossier attestent de l’importance de la stratégie de communication du laboratoire auprès des KOL, en particulier auprès du Professeur Y…, directeur de l’étude GEFAL – qui préoccupait particulièrement Novartis –, mais également des principaux chercheurs impliqués (cotes 3630, 6311, 2470, 45132, 4358, 13993, 13996, 4629, 14045, 3106 et 3107, 2567, 4494 et 3048). Par ailleurs, Novartis a également choisi d’orienter sa communication auprès des associations de patients et du grand public (cotes 6311, 14045, 1572, 4494, 3048 et 15826), ainsi que des autorités de santé (cotes 14045, 1572, 1557, 4494, 3048 et 45146). Enfin, le communiqué de presse, publié par Novartis le 14 mai 2013, après que les résultats de l’étude GEFAL ont été rendus publics (cotes 14997 et 14998, cote 2985), a été diffusé à l’ensemble de la presse médicale et pharmaceutique, tant généraliste que spécialisée et aux agences de presse grand public, en ce compris la presse santé et la presse pour les seniors (cote 2978).
790. En outre, Novartis a ciblé ses actions sur les centres d’injections connus pour utiliser Avastin en ophtalmologie, appelés « key centers ». À titre d’illustration, le document de stratégie de juillet 2009 montre que, lorsqu’un médecin déclarait utiliser Avastin, il était alors invité à une réunion régionale à l’occasion de laquelle Novartis revenait sur les éléments de sécurité et de tolérance liés à l’utilisation d’Avastin en ophtalmologie (cote 2407). De même, dans le document de présentation de la réunion « M3PH » de juin 2010, à l’occasion de laquelle Novartis a présenté sa stratégie pour Lucentis pour l’année 2011, le laboratoire planifie des actions ciblées auprès des « key centers », visant en particulier les Hospices civils de Lyon, et notamment le Professeur Y…, directeur de l’étude GEFAL, qu’il importe de « maintenir occupé » (cote 13996). Par ailleurs, le compte rendu de la réunion « LBT » (ou « Local Brand Team ») de juillet 2010 indique que les représentants de Novartis « pourront avoir une action plus proactive, mais coordonnée et ciblée sur centres utilisateurs ou proches de basculer ou sur les centres faisant partie du halo d’influence des autorités de santé (cote 4629). Ou encore, dans une présentation du 28 septembre 2010, Novartis précise que les « messages clés » doivent être diffusés en particulier aux médecins les plus orientés vers Avastin (« selected Avastin lovers », traduction libre : « des amoureux d’Avastin sélectionnés », cote 14045).
791. Ainsi, il ressort des éléments du dossier que Novartis a ciblé ses actions de communication sur les professionnels de santé représentant un enjeu stratégique.
792. En second lieu, les pièces internes de Novartis montrent que ses actions ont eu un impact sur les volumes des ventes d’Avastin, ce qui atteste de leur mise en œuvre effective et que les déclarations des médecins visées par Novartis ne suffisent pas à contredire (cf. paragraphes 837 à 888 de la présente décision).
793. Par conséquent, contrairement à ce que soutient Novartis, il est établi que celui-ci a diffusé auprès des professionnels de santé, des associations de patients, du grand public et des autorités de santé, les éléments d’une communication portant sur les risques associés à l’utilisation d’Avastin en ophtalmologie, et plus particulièrement pour le traitement de la DMLA.
♦ Le contenu du discours délivré par Novartis
794. Novartis soutient que sa communication s’est inscrite dans le cadre de l’exercice de sa liberté d’expression, protégée par l’article 10 de la CEDH, dans un débat d’intérêt général sur la santé publique, concernant les implications financières et sanitaires de l’utilisation « hors AMM » d’Avastin en ophtalmologie, en lieu et place de Lucentis, spécialité autorisée. Le laboratoire affirme à cet égard que sa communication s’est toujours appuyée sur les données factuelles scientifiques disponibles, traduisait la manifestation sincère de sa conviction au moment des faits, et a toujours été exprimée avec mesure.
795. Plus spécifiquement, Novartis soutient que ce n’est qu’à compter du moment où une autorité compétente aurait établi l’équivalence du profil bénéfice/risque entre les deux spécialités ou aurait expressément autorisé l’utilisation « hors AMM » d’Avastin dans le traitement de la DMLA, que sa communication, mettant en avant un excès de risque pouvant résulter de la prescription d’Avastin en ophtalmologie, pouvait être susceptible d’être qualifiée de trompeuse. Le laboratoire estime à cet égard que les études scientifiques publiées à l’époque des faits ne constituaient pas une base suffisante pour considérer que la mention de l’existence d’un risque lié à l’utilisation d’Avastin en comparaison avec Lucentis serait trompeuse et ce, d’autant plus dans un contexte réglementaire où, à la différence de la situation réglementaire en cause dans l’affaire italienne, l’usage d’Avastin en ophtalmologie n’était pas remboursé par la sécurité sociale et les autorités de santé déconseillaient cet usage, voire même l’avaient interdit.
796. Toutefois, les arguments de Novartis ne sauraient convaincre, pour les raisons suivantes.
797. À titre liminaire, il convient de souligner que ce n’est pas le principe même de la communication de Novartis qui est mis en cause dans le grief notifié, mais la teneur spécifique de son discours en l’espèce, en considération du contexte dans lequel il s’est inscrit.
798. À cet égard, d’une part, la communication de Novartis ne s’inscrivait pas dans le cadre de ses obligations de pharmacovigilance. Sur ce point, la Cour de justice a jugé, dans son arrêt précité Roche/Novartis, que « les exigences de pharmacovigilance pouvant impliquer des démarches telles que la diffusion auprès des professionnels de la santé et du grand public d’informations relatives aux risques liés à l’utilisation hors AMM d’un médicament, de même que l’ouverture d’une procédure auprès de l’EMA en vue d’inclure de telles informations dans le résumé des caractéristiques du produit, reposent, ainsi qu’il ressort des dispositions mentionnées aux points 82 à 87 du présent arrêt, sur le seul titulaire de l’AMM dudit médicament et non pas sur une autre entreprise commercialisant un médicament concurrent, couvert par une AMM distincte » (arrêt du 23 janvier 2018, Hoffmann-Laroche, C-179/16, point 91). En l’espèce, l’obligation de communiquer sur les risques liés à l’utilisation d’Avastin « hors AMM » incombait donc au seul titulaire de l’AMM, donc à Roche, et non à Novartis.
799. D’autre part, l’examen du contenu du discours de Novartis, à la lumière du contexte dans lequel il s’est inscrit, montre que celui-ci n’était pas motivé par des considérations de santé publique, mais qu’il s’inscrivait dans le cadre d’une manœuvre anticoncurrentielle, visant à empêcher l’utilisation d’Avastin « hors AMM » en ophtalmologie, au profit de son propre produit, Lucentis, comme cela ressort des développements ci-après.
800. En effet, en premier lieu, les documents présents au dossier montrent que le discours de Novartis constituait un discours commercial, qui n’était aucunement motivé par des considérations scientifiques ou sanitaires. Conscient du peu de données disponibles permettant de différencier Lucentis d’Avastin, Novartis a, par ce discours, exagéré les incertitudes concernant l’usage « hors AMM » d’Avastin.
801. Par exemple, dans le document intitulé « Lucentis C4 Plan 2009-2014 wAMD [wet age-related macular degeneration] », de juillet 2009, Novartis constatait qu’un des principaux enjeux pour Lucentis était l’absence de différenciation prouvée cliniquement et le prix élevé du médicament commercialisé par Novartis (« Key issues (…) No proven clinical differentiation between Lucentis & Avastin, and Lucentis price perceived as high », cote 2376). Novartis entendait par conséquent se préparer au cas où l’étude GEFAL démontrerait la non-infériorité d’Avastin par rapport à Lucentis en termes d’efficacité afin d’en minimiser l’impact (« GEFAL study has started (…). We need to be prepared in case of proven non-inferiority in terms of efficacy to minimize impact », cote 2377).
802. Il relevait par ailleurs qu’il était probable qu’une majorité de médecins français anticipaient que les études de comparaison en cours – et notamment GEFAL – allaient démontrer l’équivalence d’Avastin et de Lucentis, non seulement en termes d’efficacité, mais également de sécurité, et qu’une majorité des médecins anticipait déjà une équivalence d’efficacité et de sécurité. Novartis en déduisait la nécessité de mettre en place rapidement des actions pour différencier Lucentis par rapport à Avastin (« Head to Head trials will likely show non inferiority. Actions to differenciate Lucentis vs Avastin should be quickly set up as physicians already anticipate similar safety and efficacy (56 %) », cote 2392).
803. En second lieu, le discours diffusé par Novartis n’était pas fondé sur une base factuelle suffisante et n’a pas été exprimé avec suffisamment de mesure.
804. Premièrement, Novartis a diffusé un discours suggérant qu’il pouvait exister un risque d’effets systémiques supplémentaires liés aux propriétés d’Avastin, différentes de celles de Lucentis. Dans ce cadre, Novartis a insisté sur le profil de sûreté de Lucentis, garanti par la réalisation des études cliniques préalables à son AMM, en l’opposant à l’absence de données certaines sur l’efficacité et la sécurité d’Avastin en ophtalmologie.
805. Comme le souligne Novartis dans ses observations, il est exact que le laboratoire n’a fait qu’évoquer la possibilité d’un tel lien entre les différences entre les deux molécules et les effets indésirables constatés avec Avastin, sans l’affirmer de façon certaine. Il n’en demeure pas moins que, d’une part, la présentation que Novartis a faite du lien entre les effets indésirables systémiques constatés et les propriétés d’Avastin ne reposait pas sur une base factuelle suffisante et que, d’autre part, associée à la présentation des garanties attribuées à Lucentis en termes d’efficacité et de tolérance, un tel discours n’était pas exprimé avec suffisamment de mesure, compte tenu du contexte scientifique dans lequel il s’inscrivait.
806. En effet, d’une part, Novartis a insisté sur les différences moléculaires et de pharmacodynamique entre le ranibizumab (Lucentis) et le bevacizumab (Avastin), de manière à faire apparaître un lien entre celles-ci et les effets indésirables systémiques plus importants pour Avastin, identifiés par les études scientifiques. Pourtant, premièrement, les résultats des études scientifiques ne permettaient pas d’affirmer de façon certaine quelle était la cause de l’existence d’effets indésirables systémiques plus importants pour Avastin (cf. paragraphes 94 à 128 de la présente décision) et, deuxièmement, le CHMP – organe de santé compétent pour examiner la portée de ces études – a, quant à lui, interprété les résultats de ces études, dès 2012, comme « rassurantes » et n’apportant « pas de preuve que le bevacizumab [Avastin] est moins sûr que le ranibizumab [Lucentis] s’agissant des effets systémiques » (cf. paragraphes 130 à 148 de la présente décision).
807. À titre d’illustration, dans le « Lucentis Defend plan » d’avril 2011, Novartis souligne : « Serum concentration differences could suggest systemic AE [adverse effects] » (cote 14105). Pourtant, les résultats des études Curtis et Gower, publiés respectivement en octobre 2010 et en avril 2011, indiquaient que la démonstration de l’existence et de la cause d’effets indésirables supplémentaires pour Avastin restait à déterminer (cf. paragraphes 98 à 103 de la présente décision).
808. De même, dans un document intitulé « CATT, les Messages clés », commentant les résultats de l’étude CATT rendus publics en mai 2011 et juillet 2012, Novartis indiquait : « Les écarts de tolérance observés dans CATT entre Avastin et Lucentis sont probablement liés aux différences de structure moléculaire et d'exposition systémique consécutive aux injections intravitréennes » (cote 3103). Pourtant, l’équipe de l’étude CATT a estimé que la cause du taux plus élevé d’événements indésirables pour Avastin était incertaine (cf. paragraphes 105 à 110 de la présente décision). En ce sens, comme souligné ci-dessus, le CHMP a indiqué, dans son avis de juillet 2012 faisant suite à la modification du RCP d’Avastin, que les résultats de cette étude (et ceux de l’étude IVAN) n’apportaient pas la preuve qu’Avastin avait un profil de sécurité systémique moindre que Lucentis (cf. paragraphes 132 à 139 de la présente décision).
809. Au surplus, un document interne de Novartis de 2013 montre qu’à cette date, il n’existait pas de lien démontré entre les caractéristiques pharmacologiques respectives des deux produits et l’existence d’effets indésirables systémiques plus importants pour Avastin (« IVAN Steering Committee member believes Avastin causes SAEs previously unidentified as VEGF related », cote 4649). Dans le même sens, le Professeur Y… a indiqué en audition, au sujet de la méta-analyse incluant CATT, IVAN et GEFAL : « Dans la méta-analyse, on a inclus GATT, IVAN, MANTA et GEFAL. Ce n’étaient que des résultats à un an. Nous avons trouvé plus d’effets indésirables graves systémiques avec l’Avastin. Le problème, c’est qu’on n’a pas su identifier pourquoi. Pas de différence sur la mort, sur les APTC (là où on attend la molécule) ou sur les effets gastro-intestinaux, pas de différence non plus » (cote 16623). D’ailleurs, dès 2009, Novartis reconnaissait dans un document de stratégie interne, que l’un des principaux problèmes de Lucentis (« Lucentis Prioritized Key Issues ») était l’absence de différenciation prouvée cliniquement avec Avastin (« Key issues (…)
No proven clinical differentiation between Lucentis & Avastin, and Lucentis’ price perceived as high (…) », cote 2376).
810. D’autre part, Novartis a insisté sur les points de différenciation entre Avastin et Lucentis, en soulignant plus particulièrement les garanties de Lucentis. À titre d’illustration, dans un document de mars 2008, Novartis oppose la sécurité d’usage de Lucentis aux incertitudes liées à l’utilisation d’Avastin (« Lucentis is specifically designed and proven safe for use in the human eye (…) No proper data are available on safety or serious side effects with Avastin use in wet AMD (…) its safety as a treatment for wet AMD is unknown», cote 3612). Novartis fait de même, dans un document intitulé « Elevator Speech » de juin 2010 (« Avastin is different from Lucentis (…) Avastin is uncertain (…) Avastin is unapproved (…) NVS [Novartis] is a true partner for ophtalmology », traduction libre : « Avastin est différent de Lucentis (…) Avastin est incertain (…) Avastin n’est pas autorisé (…) NVS [Novartis] est un vrai partenaire pour l’ophtalmologie », cote 13988 ; voir également cote 3473). Au début de l’année 2012, Novartis focalise toujours son argumentaire sur la potentialité d’effets indésirables plus importants avec Avastin, en comparaison avec les garanties de Lucentis (« Safety concerns: there is an emerging body of evidence indicating that the risk of ATEs [adverse effects] may be higher with unlicensed Avastin compared to Lucentis (…) Lucentis Licensed (…) Designed for the eye (…) Lucentis has well characterized safety profile », traduction libre : « Préoccupations de sécurité : il y a un ensemble de preuves qui fait surface indiquant que les risques d’événements indésirables pourraient être supérieurs avec l’utilisation ‘hors AMM’ d’Avastin en comparaison avec Lucentis (…) Lucentis a une AMM (…) Lucentis est conçu pour une utilisation dans l’œil (…) Lucentis a un profil de sécurité établi », cote 1572). Ou encore, dans le document «Messages clés » mis à jour en mars 2013, Novartis insiste sur l’existence de médicaments validés par les autorités publiques, en opposition aux incertitudes qui demeurent sur la tolérance et la sécurité d’Avastin (cote 2983).
811. Par conséquent, il ressort des pièces du dossier que la présentation par Novartis, dans sa communication, des causes potentielles des risques liés à l’utilisation d’Avastin, opposées aux certitudes sur l’innocuité de Lucentis, n’a pas été exprimée avec suffisamment de mesure, compte tenu du contexte scientifique dans lequel son discours s’inscrivait.
812. Deuxièmement, Novartis a effectué une présentation sélective et biaisée des résultats des études scientifiques comparant l’efficacité et la sécurité de l’utilisation d’Avastin et de Lucentis en ophtalmologie.
813. En effet, il ressort des pièces du dossier que Novartis a sélectionné les informations allant dans le sens de son argumentaire, tendant à insister sur les risques liés à l’utilisation « hors AMM » d’Avastin en ophtalmologie, en comparaison avec la sécurité établie de Lucentis.
814. Novartis a ainsi insisté sur les limites méthodologiques des études scientifiques uniquement lorsqu’il présentait des résultats qui n’étaient pas défavorables à Avastin. À l’inverse, lorsque ces résultats permettaient de mettre en avant des risques liés à l’utilisation d’Avastin en ophtalmologie, Novartis passait sous silence les limites de ces études.
815. À titre d’illustration de cette démarche, dans une présentation intitulée « Head-to-head trials of Lucentis versus Avastin », Novartis soulignait les limites des études CATT et IVAN, pour en tirer argument de ce que celles-ci ne permettraient pas d’obtenir des conclusions sérieuses sur l’efficacité et la sécurité d’Avastin en ophtalmologie (« it is unlikely that any of the current ongoing head-to-head trials are powered for safety larger studies powered to detect potentially significant systemic adverse events may be required. Possible Bias for non-inferiority (…) Limited scientific value and interpretability », traduction libre : « Il est peu probable que les études en cours soient probantes pour la sécurité de plus larges études réalisées pour détecter les éventuels événements indésirables systémiques graves seront probablement requises. Il y a un biais probable sur la non-infériorité (…) valeur scientifique et interprétabilité limitée », cote 14976). Pourtant, Novartis a exploité, dans de nombreux documents de communication et de manière proactive (« proactive positionning of CATT and IVAN results », cote 2567), une partie des résultats de ces études (et de l’étude CATT en particulier), après leur publication, sans en mentionner les limites précédemment exposées, ni même les conclusions générales qui étaient pourtant « rassurantes », selon les termes du CHMP dans son avis susvisé (cf. paragraphes
132 à 139 de la présente décision), comme :
- dans les « Messages clés » mis à jour après la publication des résultats de l’étude CATT à deux ans, en juillet 2012, qui indiquent que « les résultats de l'étude CATT à 2 ans confirment les très sérieux doutes de tolérance pour Avastin en ophtalmologie » (cote 1577) ;
- dans le document « CATT, les Messages clés » susvisé – commentant les résultats de l’étude CATT rendus publics en mai 2011 et juillet 2012 –, qui précise : « Dans l'étude CATT, un plus grand nombre de décès et une augmentation statistiquement significative du risque d'accidents systémiques sérieux nécessitant une hospitalisation ont été observés avec Avastin comparé à Lucentis (29 % de risques supplémentaire avec Avastin pour les accidents systémiques sérieux. Ce signal en termes de tolérance est cohérent avec l'ensemble des résultats obtenus dans de précédentes études cliniques, notamment les larges analyses Medicare, indiquant que les risques de mortalité et d'accident vasculaire et cérébral sont plus élevés avec Avastin comparé à Lucentis (…) » (cote 3103) ;
- ou encore, dans une présentation, intitulée « Lucentis H2H Action Plan », datée d’avril 2013, qui souligne : « Pooled CATT & IVAN study showed significant increase in serious adverse events » (traduction libre : « les études combinées CATT et IVA ont montré une augmentation importante des effets indésirables graves », cote 14841).
816. À l’inverse, Novartis, qui avait conscience de ce que les résultats de l’étude GEFAL auraient un impact certain sur l’utilisation d’Avastin en ophtalmologie, a insisté sur les limites méthodologiques de cette étude, pour en réduire la portée.
817. En effet, dans un document de juillet 2008, Novartis soulignait la menace que l’étude GEFAL représentait pour le laboratoire : « Avastin threat is concrete (Gefal Study) », cote 45083). À ce titre, Novartis préconisait, dès cette date, les actions à mettre en œuvre pour minimiser l’impact de cette étude (« Minimize H2H studies (Gefal) impact : Secure Lucentis price, closely monitor the implementation, communicate on insufficiencies », cote 2440). Ainsi, dès juillet 2008, Novartis envisageait de remettre en cause cette étude et de communiquer sur ses « insuffisances » (« challenge Gefal design and communicate on insufficiencies », cote 2470).
818. Aussi, Novartis a axé sa communication autour de l’étude GEFAL, en insistant sur ses limites méthodologiques, pour en minimiser l’impact. Plus spécifiquement, dans un document intitulé « Messages clés Lucentis / Avastin », mis à jour en mars 2013, afin de préparer les équipes commerciales à la publication prochaine des résultats de l’étude GEFAL, Novartis indique : « L'étude française GEFAL (Groupe d'Etude Français Avastin vs Lucentis) n'a pas été dimensionnée pour lever définitivement les incertitudes sur la tolérance d'Avastin » (cote 2983). De même, dans un document de « Questions / Réponses » de juin 2013, mis à jour après la communication du Professeur Y… sur les résultats de l’étude GEFAL en mai 2013, Novartis propose d’apporter, à la question « Aujourd’hui, avec l’étude française GEFAL en plus, il y a un niveau de preuve suffisant pour utiliser Avastin en toute tranquillité dans la DMLA », la réponse suivante : « Au même titre que CATT (1206 patients) et IVAN (610 patients), l’étude GEFAL n’apporte pas de preuve suffisante pour conclure sur le profil de sécurité d’Avastin dans la DMLA », cote 2515).
819. Sur ces points, Novartis soutient notamment que les résultats des études scientifiques présentés dans sa communication étaient exacts et, qu’au surplus, il a toujours mentionné les sources de ses affirmations, permettant à ses interlocuteurs d’en vérifier la pertinence. Par ailleurs, Novartis affirme qu’il était en droit de revenir dans sa communication sur les limites méthodologiques de ces études scientifiques.
820. Néanmoins, s’il n’est pas contestable que, comme le soutient Novartis dans ses observations, les informations relayées par ses soins concernant les résultats des études scientifiques sont exactes, il n’en demeure pas moins que Novartis a sélectionné les seules informations allant dans le sens de son argumentaire, tendant à insister sur les risques liés à l’utilisation « hors AMM » d’Avastin en ophtalmologie, en comparaison avec la sécurité établie de Lucentis.
821. Au surplus, comme souligné aux paragraphes 800 et 805 ci-dessus, cette présentation sélective des résultats des études scientifiques était associée à un discours insistant particulièrement sur l’efficacité garantie de Lucentis.
822. Par conséquent, d’une part, en sortant les résultats de l’étude CATT de leur contexte et, d’autre part, en ne soulignant que les limites méthodologiques de l’étude GEFAL, sans en présenter les conclusions, Novartis a procédé à une présentation parcellaire et décontextualisée des résultats des études scientifiques disponibles.
823. Troisièmement, Novartis a également fait une présentation sélective, et par suite trompeuse, des modifications opérées sur le RCP d’Avastin et de Lucentis. Sa communication sur ce point suggérait en effet, d’une part, que l’autorité européenne de santé avait décidé d’informer le public de l’existence d’effets indésirables systémiques uniquement pour Avastin, alors pourtant que le CHMP avait estimé qu’il ne convenait pas de faire une distinction entre les différents anti-VEGF et, d’autre part, que seule l’utilisation d’Avastin entraînerait des effets indésirables oculaires, alors que le RCP de Lucentis contenait également un avertissement sur les effets oculaires liés aux injections de Lucentis.
824. De fait, s’agissant des effets indésirables systémiques, en juillet 2012, le CHMP a estimé qu’il était nécessaire de mettre à jour la section 4.4 « Mises en gardes spéciales et précaution d’emploi » du RCP d’Avastin, afin d’insérer, notamment, un paragraphe sur l’existence d’effets systémiques indésirables liés à l’utilisation des anti-VEGF en intra-vitréen (cf. paragraphes 132 à 139 de la présente décision). Six mois plus tard, en janvier 2013, le CHMP a préconisé d’intégrer exactement la même mention dans la section 4.4 « Mises en gardes spéciales et précaution d’emploi » du RCP de Lucentis (cf. paragraphes 140 à 148 de la présente décision).
825. Pourtant, Novartis a communiqué exclusivement au cours de l’été 2012 autour de la modification du RCP d’Avastin, qui n’est pourtant pas le produit qu’elle commercialise, sans s’exprimer, à aucun moment, sur l’ajout de la même mention dans le RCP de son propre produit Lucentis, qui a pourtant eu lieu peu de temps après.
826. Dans une présentation de janvier 2012, Novartis prévoit de mettre à profit le changement probable à venir du RCP d’Avastin comme un « levier » : « leverage likely upcoming label change of Avastin (…) » (cote 1566). Par ailleurs, un document intitulé « Counter Avastin materials » (traduction libre : « Eléments anti-Avastin »), de février 2012, prévoit que le siège de Novartis devra transmettre aux filiales des informations sur ce point (« Global (…) Avastin label change (systemic ocular AEs [adverse effects]) », cote 2567) lorsque la modification sera faite.
827. Novartis soutient sur ce point qu’il ne saurait lui être reproché de ne pas avoir communiqué sur le changement de RCP de Lucentis, dès lors que cette modification a été préconisée par le CHMP en janvier 2013 et qu’elle n’a, postérieurement à cette date, plus communiqué sur la modification du RCP d’Avastin. Le laboratoire avait à ce sujet souligné, en réponse à une demande d’informations, ne pas avoir communiqué au sujet de la modification du RCP de son propre produit « dans la mesure où l’Agence européenne des médicaments ne l’a pas demandé. Toutefois, tous nos supports promotionnels mentionnant le RCP ont évidemment été mis à jour pour refléter ce changement » (cote 45014). Il a produit en outre, dans ses observations en réponse au rapport, des exemples de documents promotionnels contenant explicitement la mention des effets indésirables systémiques pouvant résulter de l’utilisation de Lucentis.
828. Mais, d’une part, figurent au dossier des pièces attestant du fait que Novartis a communiqué sur le seul changement de RCP d’Avastin, après la modification du RCP de Lucentis, par opposition à celui de Lucentis, où il n’y a pas eu de communication proactive, comme en témoignent l’« argumentaire détaillé », daté de juin 2013 (cote 2518) et celui de mars 2014 (cote 15787).
829. S’il est exact que, comme le soutient Novartis dans ses observations, ces deux derniers documents ne reviennent que sur les effets indésirables oculaires ajoutés au RCP d’Avastin, Novartis omet de préciser que le RCP de Lucentis contient également un avertissement sur les effets oculaires liés aux injections. En effet, ce RCP précise, dans la section 4.4 « Mises en garde spéciales et précautions d’emploi » que « Les injections intravitréennes, y compris celles de Lucentis, ont été associées à des endophtalmies, des inflammations intraoculaires, des décollements rhegmatogènes de la rétine, des déchirures de la rétine et des cataractes traumatiques iatrogènes (voir rubrique 4.8) ». La section 4.8 « Effets indésirables » précise également que « La majorité des effets indésirables rapportés après l’administration de Lucentis sont liés à la procédure d’injection intravitréenne. Les effets indésirables oculaires les plus fréquemment rapportés après l’injection de Lucentis sont : des douleurs oculaires, des hyperhémies oculaires, des augmentations de la pression intraoculaire, des hyalites, des décollements du vitré, des hémorragies rétiniennes, des troubles visuels, des corps flottants vitréens, des hémorragies conjonctivales, des irritations oculaires, des sensations de corps étranger dans l'œil, des sécrétions lacrymales accrues, des blépharites, des sécheresses oculaires et des prurits oculaires ». Or l’« argumentaire détaillé » susvisé évoque des effets oculaires communs à ceux présentés ci-dessus : « Les effets indésirables liés à l’utilisation d'Avastin en intra-vitréen, ont conduit à une modification de la notice et du RCP européens où figurent désormais les mises en garde spéciales et précautions d'emploi suivantes : des événements indésirables ont été rapportés concernant l'utilisation non approuvée d'Avastin en intra-vitréen, Ces effets indésirables incluent des endophtalmies Infectieuses, des inflammations intraoculaires (telles que endophtalmies stériles, uvéites, inflammations du vitré), des décollements rétiniens, des déchirements de l'épithélium pigmentaire, des hausses de la pression intra-oculaire. Des hémorragies intraoculaire ou du vitré » (cotes 2518 et 15787).
830. D’autre part, et surtout, même si l’EMA ne lui a pas imposé de communiquer sur la modification du RCP de Lucentis, il aurait été cohérent que la laboratoire Novartis le fasse, puisqu’il s’agissait de son propre produit et qu’il avait largement communiqué sur le changement de RCP d’Avastin, produit qu’il ne commercialisait pas. Dès lors, l’absence de communication sur le changement de RCP de Lucentis constitue un indice fort du fait que cette communication sur le changement de RCP d’Avastin n’était pas motivée par des considérations de santé publique, mais bien par la seule visée anticoncurrentielle de limiter l’usage d’Avastin pour traiter la DMLA. Les exemples de documents promotionnels produits par Novartis ne sont, en tout état de cause, pas probants, car il s’agit de « fiches posologie », correspondant au document standard, généralement remis par les laboratoires pharmaceutiques aux professionnels de santé, afin de respecter leur obligation d’information prévue à l’article R. 5122-8 du code de la santé publique, notamment sur la posologie ou les effets indésirables du produit concerné. En revanche, Novartis n’apporte pas la preuve que, dans le cadre de sa communication, le laboratoire aurait appelé l’attention des professionnels de santé sur la modification du RCP de Lucentis préconisée par le CHMP en janvier 2013, alors qu’elle a insisté sur celle opérée, peu de temps auparavant, pour Avastin.
831. D’ailleurs, le CHMP a souligné, dans son avis du 17 janvier 2013, d’une part, que : « les données [des études de pharmacocinétique et pharmacodynamique] ne sont, à elles seules, pas suffisantes pour justifier un avertissement différencié qui donnerait l’impression que Lucentis serait plus sûr que les autres traitements par anti-VEGF concernant les effets indésirables systémiques » et, d’autre part, que « les résultats des études CATT et IVAN ont déjà été étudiés par le CHMP dans une procédure antérieure, arrivant à la conclusion qu’il n’y avait pas de preuve d’une différence dans le profil de sécurité systémique du ranibizumab [Lucentis] et du bevacizumab [Avastin] »), pour en conclure qu’il ne convenait pas d’ajouter un « avertissement qui donnerait l’impression que le ranibizumab [Lucentis] est plus sûr que les autres traitements anti-VEGF intravitréens, s’agissant des effets indésirables systémiques » (cf. paragraphes 145 et 146 de la présente décision).
832. Par conséquent, en communiquant uniquement sur le changement de RCP d’Avastin, Novartis a effectué une présentation suggérant qu’il s’agissait d’effets indésirables détectés uniquement pour ce produit, alors même que le CHMP avait estimé qu’il ne convenait pas de faire une distinction entre les différents anti-VEGF.
833. Quatrièmement, Novartis a insisté sur la responsabilité civile et pénale des professionnels de santé qui prescriraient Avastin « hors AMM ».
834. À titre d’illustration, dans les documents de préparation du « media training », en vue d’une conférence au Club francophone des spécialistes de la rétine, début 2008, Novartis indique : « Par conséquent, les médecins qui utilisent Avastin en dehors de son indication le font à titre expérimental et engagent leur responsabilité civile en cas de complication » (cote 3593). Le même message apparaît dans les messages clés et réponses à objections présentés lors de la réunion « Advocacy Lucentis » en mai 2010 (« le médecin peut engager sa responsabilité civile, pénale, administrative et disciplinaire si la prescription hors AMM d'un produit n'est pas justifiée », cote 2138) ou encore dans l’« Elevator Speech » de juin 2010 : « Le médecin et le pharmacien engagent leur responsabilité », cote 13991). La présentation intitulée « Lucentis Defend Plan » d’avril 2011 insiste plus fortement encore sur les risques juridiques en termes de responsabilité médicale pour les professionnels de santé utilisant Avastin « hors AMM », en évoquant notamment l’affaire du Mediator (« Legal issues and physician liabilities regarding off label prescriptions is also to be highlighted in a Mediator crisis context », traduction libre : « Les problématiques juridiques et les responsabilités des praticiens concernant les prescriptions ‘hors AMM’ doivent aussi être mises en avant dans un contexte de crise du Médiator » cote 14110). Il en va de même du document de « Questions / Réponses » de juin 2013, qui contient de nombreux arguments relatifs aux risques en termes de responsabilités civile, pénale et disciplinaire, pour un médecin qui prescrit Avastin « hors AMM », alors qu’il existe une alternative thérapeutique disposant d’une AMM, et dans un contexte de récents scandales sanitaires (« le médecin engage sa responsabilité civile, pénale et disciplinaire s’il fait courir un risque injustifié au patient tandis qu’il existe une alternative thérapeutique disposant de l’AMM dans cette indication », cote 2512 ; voir également cote 2519 et 2523).
835. Par conséquent, contrairement à ce que soutient Novartis, le laboratoire ne s’est pas contenté d’évoquer les différences objectives entre Lucentis et Avastin, ni de rappeler fidèlement le contexte scientifique relatif à l’utilisation d’Avastin. Au contraire, Novartis a présenté à ses interlocuteurs un ensemble d’éléments de comparaison d’Avastin et Lucentis, en s’appuyant notamment sur une présentation sélective et biaisée des études scientifiques disponibles et des modifications du RCP d’Avastin sur les effets secondaires indésirables, ou encore en insistant sur les questions de responsabilité des professionnels de santé prescrivant « hors AMM », en vue d’exagérer les risques liés à l’utilisation d’Avastin « hors AMM » pour le traitement de la DMLA et, plus généralement, en ophtalmologie.
♦ Conclusion
836. Il ressort de ce qui précède que Novartis a diffusé un discours dénigrant, en exagérant, de manière injustifiée, les risques liés à l’utilisation d’Avastin « hors AMM » pour le traitement de la DMLA, et plus généralement en ophtalmologie, en comparaison avec la sécurité et la tolérance de Lucentis pour un même usage.
Sur les effets de la communication de Novartis
837. Il convient à ce stade d’examiner les effets, potentiels ou avérés, attachés au discours véhiculé par Novartis auprès des professionnels de santé, des associations de patients, du grand public et des autorités de santé, sur le marché français du traitement de la DMLA exsudative et les marchés connexes français des autres pathologies oculaires (OMD, OBVR, OVCR, ou baisse visuelle due à une NVC).
838. Il ressort des éléments du dossier que les pratiques en cause ont été de nature à avoir, et ont eu, des effets, d’une part, sur les volumes des ventes, et d’autre part, sur les prix des spécialités concernées.
♦ En ce qui concerne l’effet sur les volumes
839. Compte tenu du contexte dans lequel le discours de Novartis a été diffusé (cf. paragraphes 761 à 768 de la présente décision), la campagne de communication du laboratoire a été de nature à provoquer l’inquiétude des professionnels de santé, des associations de patients, du grand public et des autorités de santé.
840. En effet, la tenue d’un discours dénigrant à l’encontre d’un produit concurrent, comme celui diffusé par Novartis à l’encontre d’Avastin, est susceptible d’entraver son accès au marché, a fortiori dans le secteur des médicaments, lorsqu’il met en cause l’efficacité ou la sécurité d’utilisation de ce médicament, compte tenu de l’aversion marquée des professionnels de santé pour le risque, dans un contexte caractérisé par la judiciarisation croissante des questions de santé.
841. Ainsi, toute contestation, fondée ou non, de la sécurité d’utilisation d’un médicament, associée à un discours insistant sur la responsabilité susceptible d’être engagée par les médecins le prescrivant, conduit quasi-inéluctablement à freiner son utilisation, ce qu’aucun laboratoire pharmaceutique ne peut ignorer.
842. Plus spécifiquement, en l’espèce, toute appréciation négative à l’égard d’Avastin en ophtalmologie était inévitablement de nature à provoquer l’inquiétude des professionnels de santé (médecins) et les autorités de santé, attentifs à tout élément permettant d’assurer la sécurité des patients. De même, un tel discours dirigé vers des associations de patients ou le grand public, ne disposant pas des connaissances scientifiques nécessaires pour en apprécier le bien-fondé, est également de nature à provoquer l’inquiétude des patients.
843. Un tel effet est en outre renforcé par les éléments suivants. D’une part, Novartis jouit d’une réputation tout à fait particulière dans le domaine de l’ophtalmologie, celui-ci se présentant d’ailleurs comme le laboratoire de référence en ophtalmologie. D’autre part, le discours de Novartis a été favorisé par le positionnement adopté par les autres membres de l’entité collective, Roche et Genentech ne s’étant pas opposés au discours véhiculé par Novartis, compte tenu de leur intérêt commun à favoriser les ventes de Lucentis, au détriment de celles d’Avastin.
844. Partant, toute exagération, directe ou indirecte, des risques liés à l’utilisation « hors AMM » d’Avastin en ophtalmologie, ne pouvait qu’avoir un effet sensible sur les professionnels de santé soucieux de protéger les intérêts des patients et de les inciter à privilégier Lucentis, au détriment d’Avastin.
845. Par ailleurs, il ressort des pièces du dossier que la communication a eu un impact réel et significatif sur le comportement des professionnels de santé, et donc, sur la structure du marché. Ces documents indiquent en outre que Novartis s’attachait à suivre de façon très précise et ciblée les modifications des pratiques des centres hospitaliers, résultant des pratiques.
846. En effet, d’abord, ce discours a permis de limiter l’usage « hors AMM » d’Avastin dans de nombreux établissements hospitaliers. En effet, Novartis anticipait que la concurrence d’Avastin aurait un impact sur les volumes de ventes de Lucentis. Pour limiter ce risque, Novartis a cherché, dans un premier temps, à limiter la part de marché d’Avastin en dessous de 10 % pour le traitement de la DMLA, comme en atteste le document intitulé « 2009 Retina Business Plan » de juillet 2008 (« Contain Avastin < 10% in wAMD - anticipating H2H results », cote 2447). Ainsi, au sujet de son action contre Avastin auprès des professionnels de santé, le document interne de Novartis, intitulé « Suivi Business Rétine » du 24 avril 2008, indique : « situation résolue aux 15/20, à la fondation Rothschild, à Grenoble, à Lille, à Toulon. Situation toujours bloquée à Dijon, Nantes, Cannes, Limoges, hôtel Dieu… le plus souvent pour des raisons financières (ces centres gagnent plus en utilisant l'Avastin plutôt que Lucentis) » (cote 45134), ou encore : « le nombre de centres utilisant l'Avastin a diminué (-8) grâce aux actions conjointes des délégués Rétine et des KAMs [« Key account manager » en charge des relations avec les services hospitaliers]. Reste 10 centres Avastin identifiés » (cote 3683). De même, en juin 2010, Novartis constatait les effets de la pratique visée au titre du premier grief : « Avastin usage in wAMD now limited to four main centers in France » ou encore « Avastin patient share has been steadily decreasing over the last 2 years » (cote 13977). Dans une présentation interne, intitulée « plan tactique régional Market Access », de novembre 2011, Novartis indique que, concernant le CHU de Besançon, le « résultat obtenu » est le maintien de Lucentis à l’hôpital dans l’attente des résultats de l’étude GEFAL (« Anticipation sur l’étude GEFAL : maintien de Lucentis à l’hôpital », cote 15694). Ou encore, dans une présentation établie en vue d’une réunion « MAREG » pour la Franche-Comté, le 29 juin 2012, Novartis souligne que, au regard des objectifs ainsi énoncés : « s’opposer au référencement d’AVASTIN en ophtalmo pour indications OMD et OVR » pour le centre hospitalier de Vesoul (cote 15699) ou encore de « transférer les unités d’Avastin utilisé dans ces indications en unités de Lucentis » pour le CHU de Besançon (cote 15699) –, les résultats obtenus sont les suivants : « DMLA, OMD et OVR. 100 % Lucentis » pour le CHU de Besançon (cote 15700 ; voir également cote 15707 pour le CHU de Dijon).
847. Ainsi, comme souligné au paragraphe 598 de la présente décision, dans la mesure où toutes les pathologies rétiniennes traitées par injections dans l’œil d’anti-VEGF étaient les concernées par le débat sur la comparaison des effets secondaires indésirables entre Avastin et Lucentis, les pratiques mises en œuvre par Novartis ont été de nature à affecter les ventes d’Avastin pour le traitement de la DMLA et des autres pathologies oculaires (OMD, OBVR, OBCR ou NVC).
848. Ensuite, le discours de Novartis a eu un impact sur les opinions des ophtalmologistes et sur les décisions prises par les professionnels de santé concernant l’usage d’Avastin « hors AMM ». Ainsi, le compte rendu rédigé par le directeur de la « Franchise Ophtalmologie » de Novartis (direction responsable de la commercialisation de Lucentis en France), en vue de son évaluation individuelle pour l’année 2012, mentionne, parmi ses résultats satisfaisants, le fait que 50 % des spécialistes de la rétine considèrent que la sécurité d’Avastin est inférieure à celle de Lucentis et le fait qu’aucun leader d’opinion, ni aucune association de praticiens ou de patients, ne se soit prononcé publiquement en faveur d’Avastin, à l’exception de l’hôpital public de l’Hôtel Dieu, à Paris (« 50 % of RS [retina specialists] considering Avastin safety is inferior to Lucentis. Partnership built with GefaI stakeholders. No public pro Avastin position by key physicians / physicians associations / patients associations except Hôtel Dieu (post CATT 2 or post DGS letter) » (traduction libre : « 50 % des spécialistes de la rétine considère que la sécurité d’Avastin est inférieure à celle de Lucentis. Partenariat construit avec les parties prenantes de Gefal. Il n’y a pas de prise de position publique en faveur d’Avastin par les principaux praticiens / les associations de médecins / les associations de patients, à l’exception de l’Hôtel Dieu (après CATT 2 et après la lettre de la DGS) », cotes 45127 et 45128). De même, pour l’année 2013, sont mis en avant la première communication officielle de Novartis mentionnant clairement les différences de sécurité entre Avastin et Lucentis, réalisée en collaboration avec des KOL importants, et les effets de la stratégie de Novartis dans différents rapports et articles de presse, en particulier en l’absence à cette date de publication du décret concernant la RTU (« First official Novartis written communication clearly stating Lucentis vs Avastin safety différence, in coopération with top KOLs. Awareness of these safety issues has really increased in France (cf. Gefal communications, Pr Marananchi audition to senate, CNAM report to Assemblée Nationale, general pubic media articles…) which can explain that RTU décret is still not published and that Health Authorities bodies are reorienting their actions towards a Lucentis price discussion more than an Avastin endorsement » (traduction libre : « Première communication écrite officielle de Novartis indiquant les différences entre Lucentis et Avastin en termes de sécurité, en coopération avec les meilleurs KOLs [leaders d’opinion]. L’attention sur ces questions de sécurité a réellement progressé en France (cf. communication Gefal, articles dans les médias…), ce qui peut expliquer que le décret RTU n’est toujours pas publié et que les autorités de santé réorientent leurs actions vers une discussion du prix de Lucentis, plutôt qu’une approbation d’Avastin », cotes 45125 et 45126).
849. Enfin, des résultats de sondage utilisés en interne par Novartis confirment la diminution des parts de marché d’Avastin dans le traitement de la DMLA, les limites des données de parts de marché issues de la base PMSI ayant par ailleurs été présentées supra (cf. paragraphes 631 et 632 de la présente décision). Selon ces résultats, la part de marché d’Avastin est passée de 15 % en janvier 2008 (cote 13977) à 5 % en juin 2012 (cote 4705). Pourtant, selon un autre sondage, également réalisé à la demande de Novartis en octobre 2010, une part significative (36 %) des médecins ophtalmologistes interrogés avait indiqué que les résultats des études de comparaison (l’étude CATT, dont les résultats ont été publiés en mai 2011 et juillet 2012, et l’étude GEFAL, dont les résultats ont été présentés publiquement en mai 2013 lors du congrès de l’ARVO) pouvaient les inciter à augmenter leurs prescriptions d’Avastin (cotes 4342 à 4345). Dès lors, par son discours, Novartis a fait en sorte que ces transferts de prescription n’interviennent pas.
850. Roche estime que ces données de sondage ne seraient pas représentatives de la population des ophtalmologues en France pouvant prescrire Avastin « hors AMM » pour le traitement de la DMLA et que la taille des échantillons serait également trop faible par rapport à la taille de la population totale d’ophtalmologues en France. Cependant, la population française d’ophtalmologues susceptibles de réaliser des injections pour le traitement de la DMLA est très inférieure aux 5 702 ophtalmologues mis en avant par Roche : en effet, en décembre 2009, il existait, selon Novartis, 1 050 ophtalmologues injecteurs pouvant pratiquer des injections intravitréennes (cote 14895), la répartition de ces 1 050 praticiens entre ophtalmologues hospitaliers, ophtalmologues libéraux et ophtalmologues « mixtes » n’étant, elle, pas connue. De même, contrairement à ce que soutient également l’étude économique de Roche, les médecins libéraux pouvaient utiliser Avastin s’ils exerçaient dans les cliniques privées. Enfin, si la taille des échantillons utilisés diminue effectivement la précision des estimations des parts de marché d’Avastin(40), d’une part, une nette diminution de ces intervalles de confiance peut être relevée et, d’autre part, plusieurs documents qualitatifs présentés supra confirment la diminution des parts de marché mise en évidence par ces sondages. À l’inverse, aucun document interne de Novartis ne remet en cause la pertinence de ces sondages. Au contraire, comme indiqué supra, Novartis a utilisé, pendant plusieurs années, le résultat de ces sondages pour orienter et adapter sa stratégie visant à diminuer l’utilisation « hors AMM » d’Avastin dans le traitement de la DMLA. Ainsi, comme évoqué supra, Novartis constatait les effets de la pratique visée au titre du premier grief en 2010 : « Avastin usage in wAMD now limited to four main centers in France », « # centers 10 May08 5 Janv09 4 June10 » ou encore « Avastin patient share has been steadily decreasing over the last 2 years » (cote 13977).
851. Novartis soutient, quant à elle, que les déclarations des professionnels de santé auditionnés par les services d’instruction, qui affirment ne pas avoir été influencés par la communication de Novartis, remettraient en cause l’existence d’un quelconque effet des pratiques. Par ailleurs, Novartis soutient que la faible utilisation d’Avastin par les médecins ophtalmologistes proviendrait du fait que cette spécialité ne disposait pas d’AMM en ophtalmologie et que le cadre réglementaire applicable ne les autorisait pas à prescrire Avastin « hors AMM », en présence d’une spécialité autorisée, Lucentis. Enfin, selon Novartis, la période suivant la publication de la RTU pour Avastin, en juin 2015, constitue la période de référence pour déterminer l’impact éventuel des pratiques. Or, selon les données exploitées par les mises en cause, l’utilisation d’Avastin n’a pas augmenté après l’adoption de la RTU pour Avastin, en juin 2015.
852. Toutefois, en premier lieu, il ressort des paragraphes 845 à 849 ci-dessus que les actions de communication de Novartis ont eu un impact réel sur les professionnels de santé destinataires du discours et, de fait, sur les volumes d’Avastin utilisés « hors AMM » pour le traitement de la DMLA et, plus généralement, en ophtalmologie, ce que les déclarations des médecins auditionnés ne suffisent pas à contredire.
853. En deuxième lieu, contrairement à ce que soutient Novartis, le cadre réglementaire n’interdisait pas l’utilisation d’Avastin « hors AMM » pour le traitement des pathologies oculaires, comme le traitement de la DMLA (cf. paragraphes 635 à 651 de la présente décision).
854. En troisième lieu, la période postérieure à la publication de la RTU d’Avastin n’est pas une période de référence pertinente pour apprécier les effets des pratiques. En effet, l’entrée sur le marché d’Eylea en novembre 2013, dont le prix a été fixé avec une décote de 10 % par rapport au prix de Lucentis (cf. paragraphe 887 de la présente décision), a réduit la part de marché susceptible d’être conquise par Avastin postérieurement à la RTU, d’autant que la part de marché d’Eylea a considérablement augmenté dès son entrée sur le marché (pour atteindre autour de 40 % au bout de quelques mois).
855. Dès lors, les arguments de Novartis seront écartés.
856. Il ressort de ce qui précède que le discours de Novartis a été de nature à, et a eu pour effet de limiter les prescriptions d’Avastin pour le traitement de la DMLA et, plus généralement en ophtalmologie.
♦ En ce qui concerne l’effet sur les prix
857. Le discours diffusé par Novartis a également été de nature à avoir pour effet de maintenir le prix de Lucentis à un prix supra-concurrentiel et de fixer le prix d’Eylea à un prix artificiellement élevé.
858. En effet, en premier lieu, il ressort des pièces internes de Novartis figurant au dossier que le laboratoire anticipait que la concurrence d’Avastin pourrait avoir un impact sur le prix de Lucentis. Ainsi, le discours mis en œuvre par Novartis visait non seulement à préserver ses volumes de vente, mais aussi, ainsi qu’il résulte de plusieurs documents figurant au dossier, à préserver le niveau de prix de Lucentis.
859. Avant même l’arrivée effective de Lucentis sur le marché, son niveau de prix et la comparaison avec le coût réel d’Avastin faisaient l’objet d’une attention particulière de Novartis en interne. À titre d’illustration, dès 2005, Novartis soulignait la menace que constituait Avastin pour Lucentis, en indiquant que l’une des conséquences liées à l’engouement des rétinologistes était qu’« Avastin pourrait acquérir une image de traitement ayant le meilleur rapport coût/efficacité » (cote 3811).
860. De même, lorsque les résultats des différentes études de comparaison ont commencé à être publiés, Novartis anticipait la prise en compte d’Avastin comme comparateur, en particulier au moment de la renégociation du prix à l’issue de la garantie de prix européen, ou lors de l’inscription des autres indications oculaires. À titre d’illustration, dans le document intitulé « Lucentis Defense Workshop », Novartis indique : « Goal is to create an environment that is supportive of Lucentis price & reimbursment post-H2H » (traduction libre : « L’objectif est de créer un environnement qui est favorable au prix de Lucentis et à son remboursement post-H2H », cote 4356). De même, dans le document de présentation de la réunion « M3PH » de juin 2010, Novartis estime que la publication des résultats des études de comparaison pouvait aboutir à ce que les organismes payeurs utilisent Avastin comme comparateur pour faire baisser le prix de Lucentis (« payors use the trial results to pressure NVS [Novartis] to reduce price. H2H positive result will legitimate Avastin as price comparator at time of DME negotiation /wAMD re-listing negotiation », traduction libre : « les [organismes] payeurs utilisent les résultats des études pour mettre la pression sur NVS [Novartis] pour réduire le prix. Des résultats positifs des études H2H légitimeront Avastin comme comparateur prix au moment de la négociation pour [l’indication] DME/la négociation de la réinscription du prix de liste pour la DMLA exsudative », cote 13980). Ou encore, la présentation relative à la réunion des équipes européennes de Novartis du 24 janvier 2012 montre que le groupe s’inquiétait de l’impact de l’étude GEFAL sur la renégociation du prix de Lucentis prévue pour la fin 2012 (« Local H2H study (GEFAL data release Q2/Q3) : impact on 5-year price review in Q2/Q3 », cote 45064).
861. Ainsi, en limitant les prescriptions d’Avastin et en maintenant, de ce fait, Novartis en position de quasi-monopole, le discours du laboratoire a été de nature à empêcher le CEPS de considérer Avastin comme un comparateur pertinent, susceptible de justifier une baisse du prix de Lucentis.
862. Contrairement à ce que soutient Novartis dans ses observations, il existe bien une relation de causalité entre son discours et le niveau de prix de Lucentis.
863. En effet, si aucun lien de causalité n’est susceptible d’être établi entre la fixation initiale en 2007 du prix de Lucentis et le discours de Novartis, dans la mesure où les pratiques en cause ont débuté en mars 2008, il n’en demeure pas moins que le CEPS aurait pu procéder à une baisse du prix de Lucentis pendant la période de garantie de prix européen ou, à tout le moins, au moment de la renégociation du prix à l’issue de cette période.
864. Juridiquement, d’une part, des remises pouvaient être obtenues pendant la période de garantie de prix européen, laquelle s’étendait jusqu’en 2012.
865. Comme souligné aux paragraphes 40 à 42 de la présente décision, outre les clauses de révision de prix, le prix peut être baissé, sur initiative du CEPS, dans plusieurs circonstances. En particulier, le CEPS a indiqué dans son rapport annuel pour l’année 2010 que « Des baisses pourront également se trouver justifiées par le résultat des études relatives à l’utilisation des médicaments en situation réelle conduites conformément à l’article 6 de l’accord-cadre et de façon plus générale, en cas de modification significative des données scientifiques et épidémiologiques prises en compte pour la conclusion des conventions » (page 67). De même, en 2012, le CEPS soulignait que « s’agissant des produits bénéficiant d’une garantie de prix européen, le prix et les clauses afférentes peuvent être malgré tout révisés dès lors que survient une modification des conditions qui les avaient justifiés : prix européens, variation importante des coûts de production, évaluation de la spécialité, analyse médico-économique et volumes de ventes constatés » (rapport annuel pour l’année 2012, page 100).
866. En outre, comme souligné au paragraphe 81 de la présente décision, en présence d’un comparateur, le CEPS aurait pu négocier des remises à la première boîte. En effet, le CEPS a indiqué en audition sur ce point qu’« il n’y avait pas de comparateur donc il y a eu un accord prix/volume pour maîtriser l’impact budgétaire. S’il n’y avait pas de remise à la première boite, c’est qu’il n’y avait pas de comparateur » (cote 15185).
867. D’autre part, comme souligné au paragraphe 35 de la présente décision, le CEPS peut prendre comme comparateur cliniquement pertinent un médicament utilisé « hors AMM » en pratique courante.
868. Ainsi, le CEPS pouvait considérer Avastin comme comparateur pertinent de Lucentis ou d’Eylea, même en l’absence d’autorisation administrative pour cette spécialité – soit, en d’autres termes, avant l’adoption de la RTU en juin 2015 – en considération de son utilisation « hors AMM » par de nombreux médecins ophtalmologistes.
869. Or, au début de la période infractionnelle, Avastin était fréquemment prescrit pour le traitement de la DMLA et d’autres pathologies oculaires (cf. paragraphes
65 à 67 et 173 à 180 de la présente décision), dans un contexte où il existait pourtant, au départ, peu de données scientifiques disponibles concernant sa sécurité et son efficacité (cf. paragraphe 94 de la présente décision). Dans son avis du 28 mars 2007, la HAS avait d’ailleurs déjà relevé qu’« Un autre anti-VEGF, le bévacizumab (AVASTIN), est utilisé en dehors du cadre de son AMM » (cote 49333), sans pour autant le comparer, dans son examen de l’ASMR de Lucentis, aux autres spécialités disposant d’une AMM pour le traitement de la DMLA (Macugen et Visudyne).
870. Ainsi, le CEPS avait la possibilité d’observer l’utilisation d’Avastin par les médecins ophtalmologistes et de s’appuyer sur cette pratique pour négocier une diminution du prix et ce, même si les autorités de santé demeuraient prudentes sur l’usage « hors AMM » de cette spécialité.
871. Novartis soutient toutefois que, même en l’absence des pratiques, aucune baisse de prix de Lucentis n’aurait pu être négociée par le CEPS. Il fait ainsi valoir (i) qu’il n’existe pas de prix réglementé d’Avastin pour le traitement de la DMLA pouvant servir de base de comparaison ; (ii) que les négociations avec le CEPS ne sont fondées que sur les volumes vendus, une réduction des volumes entraînant une hausse des prix et inversement, comme le montrerait la hausse du prix remisé moyen et confidentiel de Lucentis suite à l’entrée d’Eylea; (iii) que le CEPS dispose d’un pouvoir de négociation très important face aux entreprises pharmaceutiques, et Novartis en particulier, si bien qu’il pouvait, même en l’absence de comparateur, choisir de diminuer le prix de Lucentis s’il jugeait celui-ci trop élevé ; ou encore (iv) que de nombreuses baisses de prix de Lucentis avaient déjà été enregistrées depuis 2008, si bien qu’aucune baisse de prix supplémentaire n’aurait pu être obtenue.
872. Les arguments des mises en cause ne sont cependant pas probants.
873. Premièrement, si le prix d’Avastin utilisé comme traitement contre la DMLA n’était pas réglementé, cette absence n’était pas de nature à empêcher le CEPS d’intégrer l’équivalence thérapeutique d’Avastin vis-à-vis de Lucentis et son coût d’utilisation environ 30 fois moindre lors des négociations commerciales de Novartis avec le CEPS.
874. D’ailleurs, si la différence entre le prix de Lucentis et le coût du traitement par Avastin peut effectivement être relativisée du fait des surcoûts dus à l’hospitalisation, voire à d’autres facteurs (cf. Annexe C de l’étude économique de Novartis en réponse au rapport), la baisse de prix qui aurait été négociée avec Novartis n’aurait pas conduit à ce que le prix de Lucentis soit similaire au coût du traitement par Avastin.
875. En outre, les documents internes de Novartis attestent que l’utilisation d’Avastin « hors AMM » en ophtalmologie pouvait exercer une pression à la baisse significative sur le prix de Lucentis, que son prix soit réglementé ou non par le CEPS, et que cet effet sur les prix était l’une des menaces principales auxquelles les pratiques visaient à faire obstacle (cf. paragraphes 259 à 260, 291 et 359 ci-dessus).
876. Deuxièmement, si, en raison de la diminution des volumes vendus de Lucentis du fait de l’entrée sur le marché d’Eylea, le prix net moyen de Lucentis a effectivement augmenté en 2014, cela ne signifie pas pour autant que le CEPS ne tient pas compte de l’arrivée sur le marché de comparateurs pertinents pour diminuer le prix des médicaments.
877. Au contraire, comme souligné ci-dessus, dans ses rapports annuels, qui décrivent notamment le mode de fixation et d’adaptation du prix des médicaments, le CEPS fait état, à plusieurs reprises, des possibilités de diminuer le prix des médicaments indépendamment d’une hausse concomitante des volumes vendus. En particulier, les rapports d’activité du CEPS publiés au moment des pratiques rappelaient que « les prix peuvent être baissés sur l’initiative du comité » dans plusieurs circonstances : d’une part, « conformément à l’article L. 162-17-4 du code de la sécurité sociale, lorsque l’évolution des dépenses de médicaments n’est manifestement pas compatible avec le respect de l’ONDAM, mais en conformité bien entendu avec les orientations des ministres et les engagements de l’accord-cadre » et d’autre part, « des baisses pourront également se trouver justifiées par le résultat des études relatives à l’utilisation des médicaments en situation réelle conduites conformément à l’article 6 de l’accord-cadre et de façon plus générale, en cas de modification significative des données scientifiques et épidémiologiques prises en compte pour la conclusion des conventions » (voir le rapport annuel du CEPS pour l’année 2010, page 67). De même, dans le rapport d’activité pour l’année 2010, le CEPS indiquait qu’à l’occasion du renouvellement d’inscription d’une spécialité, « La baisse de prix peut également être justifiée, conformément au code de la sécurité sociale, par la mise sur le marché, postérieurement à la première inscription du médicament, de médicaments concurrents aussi efficaces et moins coûteux » (page 67).
878. Sur ce point, le CEPS a rappelé, en audition, qu’il pouvait exister pendant la période de garantie de prix européen des motifs de modifications, et notamment de baisse de prix : « Sur Lucentis il y avait un prix européen en 2007, combiné à un accord prix/volume qui a conduit à une baisse au fil de l’eau du prix net. Cet accord prix/volume est intervenu en même temps que la fixation du prix. Tant qu’il n’y a pas de motif de modification du prix l’accord est resté appliqué » (cote 15185). S’il n’a pas précisé la nature de ces motifs, il a néanmoins souligné que le prix net d’un médicament est un levier permettant de contrôler le coût supporté par l’assurance maladie, indépendamment du prix de liste garanti par la comparaison européenne : « La réflexion sur l’impact budgétaire total existe notamment pour les maladies rares. L’objectif est de contrôler le coût total sans se préoccuper du prix de liste » (cote 15185).
879. Par ailleurs, les mises en cause ont elles-mêmes rappelé que le CEPS aurait pu renégocier à la baisse le prix de Lucentis, sans qu’Avastin soit un comparateur nécessairement officiel au sens de la HAS.
880. Ainsi, le coût d’Avastin (environ 30 euros) étant environ 30 fois moins élevé que ceux de Lucentis (entre 1 161 euros HT en 2007 et 790 euros HT en 2013) et d’Eylea (dont le prix facial a été fixé à partir de celui de Lucentis, avec une décote de 10 %, cf. paragraphe 887 de la présente décision), le CEPS aurait pu, en l’absence des pratiques, s’y référer pour renégocier le prix de Lucentis.
881. Troisièmement, le pouvoir de négociation du CEPS est nécessairement fortement amoindri lorsqu’il est confronté à un médicament en monopole pour le traitement d’une maladie. À l’inverse, ce pouvoir de négociation peut plus aisément être mis en œuvre lorsque le CEPS est en mesure de mettre en concurrence des traitements.
882. Ainsi, en l’espèce, le CEPS aurait pu, en l’absence des pratiques, renégocier à la baisse le prix de Lucentis, en s’appuyant sur une pratique médicale effective d’usage d’Avastin en ophtalmologie.
883. Quatrièmement, s’agissant des baisses de prix de Lucentis enregistrées depuis 2008, elles sont liées à d’autres facteurs que l’apparition d’un comparateur pertinent, comme la hausse des volumes vendus par exemple, le prix de remboursement d’un médicament étant fonction des volumes, selon la méthode suivie par le CEPS pour fixer les prix.
884. Ainsi, une baisse de prix supplémentaire, le cas échéant pendant la période de garantie de prix européen via des remises, aurait pu intervenir du fait de la reconnaissance d’Avastin comme comparateur. De même, s’agissant des comparaisons de prix entre médicaments et entre pays, présentées dans l’étude économique de Novartis, le fait que le prix de Lucentis décroisse plus rapidement que le prix des autres médicaments en France ou que son prix dans les autres pays européens, peut être dû à la fois au prix initial relativement élevé de Lucentis, plus élevé par exemple en France qu’en Italie ou qu’au Royaume-Uni, et au fait que les ventes de ce traitement ont plus rapidement progressé, ce qui est d’ailleurs admis par l’étude économique présentée par Novartis. De plus, les comparaisons sont réalisées sur la base des prix fabricants, c'est-à-dire sans tenir compte des rabais accordés indépendamment de la garantie de prix européenne. Par ailleurs, les régimes publics d’assurance maladie peuvent varier d’un pays à l’autre, ce qui empêche de comparer le prix de Lucentis en France et dans les autres pays. Ces comparaisons ne permettent donc pas de considérer que des baisses de prix supplémentaires n’auraient pu être obtenues en l’absence des pratiques reprochées.
885. En définitive, les pratiques des mises en cause ont donc bien été de nature à faire obstacle à une baisse de prix du Lucentis. En effet, dans la mesure où il a eu pour effet de limiter les prescriptions d’Avastin pour le traitement de la DMLA et, plus généralement en ophtalmologie (cf. paragraphes 839 à 856 ci-dessus), le discours de Novartis a également pu avoir pour effet de reporter la date à laquelle le CEPS a pu considérer que, compte tenu de son utilisation courante par les médecins ophtalmologistes, Avastin devait être considéré comme un comparateur dans le traitement de la DMLA et pour les autres indications oculaires. Ce n’est en effet qu’en octobre 2017 que cette spécialité a été retenue comme comparateur de Lucentis (cf. paragraphe 82 de la présente décision).
886. En second lieu, les pratiques ont également pu avoir un effet sur la fixation du prix d’Eylea, arrivé sur le marché en 2013. En effet, si le prix de Lucentis avait diminué avant l’entrée d’Eylea sur le marché, le prix de ce dernier aurait pu être fixé à un niveau inférieur, puisqu’il a été fixé au niveau du prix de liste de Lucentis, minoré d’une décote de 10 %. En outre, dans l’hypothèse où Avastin aurait été considéré comme un comparateur direct, le prix d’Eylea aurait également pu tenir compte du coût effectif de traitement avec cette spécialité.
887. En effet, lors de son audition, le CEPS a ainsi expliqué : « [le prix d’Eylea] a été fixé avec une décote de 10 %, conformément à la doctrine sur une ASMR V » (soulignement ajouté, cote 15187). Or, concernant les médicaments « me-too », c’est-à-dire sans ASMR (ASMR V), commercialisés par une entreprise concurrente, l’objectif du CEPS est « d’obtenir l’économie la plus importante possible » (rapport annuel du CEPS pour l’année 2012, page 97).
888. Dès lors, le discours de Novartis est susceptible d’avoir eu pour effet le maintien artificiel de Lucentis, puis de Lucentis et d’Eylea, à un prix supérieur à celui résultant d’une concurrence non faussée.
Sur le lien entre le discours dénigrant et la position dominante collective
889. Novartis considère que le lien entre le discours qui lui est reproché et la position dominante collective n’est pas démontré.
890. La pratique en cause a pu être mise en œuvre par Novartis, compte tenu de sa particulière notoriété dans le domaine de l’ophtalmologie, ainsi que du positionnement adopté par les autres membres de l’entité collective et de l’asymétrie d’information entre Novartis et la demande.
891. En effet, d’une part, au moment de son arrivée sur le marché en 2007, Novartis était le seul laboratoire commercialisant un anti-VEGF en ophtalmologie, traitement qui a été considéré comme « révolutionnaire » (cf. paragraphe 623 de la présente décision). Lucentis était en effet le seul traitement dans ce domaine disposant d’une AMM, conférant ainsi à Novartis un positionnement particulier sur le marché.
892. Aussi, si, comme le souligne Novartis dans ses observations, certains praticiens avaient à son égard une position de défiance compte tenu en particulier du prix particulièrement élevé de Lucentis, il n’en demeure pas moins que sa notoriété dans le domaine de l’ophtalmologie, cumulée à des forces de ventes importantes, lui a permis de maintenir une forte présence dans les congrès et colloques, et de développer des relations proches avec les KOL du secteur. De même, les relations entretenues par Novartis avec les associations de patients (Association DMLA et Retina France) étaient directement liées au fait qu’il s’agissait du laboratoire commercialisant le médicament principalement utilisé pour traiter la DMLA exsudative.
893. D’autre part, le Tribunal de l’Union européenne a jugé, dans l’arrêt Irish Sugar précité, que « si l’existence d’une position dominante collective se déduit de la position que détiennent ensemble les entités économiques concernées sur le marché en cause, l’abus ne doit pas nécessairement être le fait de toutes les entreprises en question. Il doit seulement pouvoir être identifié comme l’une des manifestations de la détention d’une telle position dominante collective » (arrêt du Tribunal du 7 octobre 1999, Irish Sugar, T-228/97, point 66).
894. Or, comme souligné au paragraphe 843 de la présente décision, compte tenu de l’intérêt commun de Roche et Genentech à favoriser Lucentis, le laboratoire Roche ne s’est pas opposé au discours véhiculé par Novartis au détriment des ventes de son propre produit, Avastin.
895. Ainsi, la diffusion du discours de Novartis a été facilitée par le fait que Roche n’avait pas intérêt à s’opposer à ce discours et que, de fait, elle n’a pas diffusé de contre-argumentaire, en réaction à celui-ci.
896. Il existe donc un lien direct entre les pratiques en cause et la position dominante collective détenue par Genentech, Novartis et Roche.
Sur la durée des pratiques
897. Concernant le début des pratiques, la première manifestation identifiable des pratiques de Novartis visant à limiter l’usage d’Avastin en ophtalmologie est intervenue en mars 2008, avec le courriel du directeur du marketing en ophtalmologie pour l’ensemble du groupe Novartis (cf. paragraphe 243 de la présente décision).
898. Novartis soutient pourtant qu’aucun discours trompeur ne lui serait imputable jusqu’à, à tout le moins, mai 2011, date de la première étude ayant conclu à la non-infériorité d’Avastin par rapport à Lucentis (résultats de l’étude CATT à 1 an), puisqu’il existait, jusqu’à cette date, une présomption de dangerosité concernant Avastin.
899. Toutefois, comme cela ressort des paragraphes 794 à 835 ci-dessus, le laboratoire ne s’est pas contenté d’évoquer les différences objectives entre Lucentis et Avastin, ni de rappeler fidèlement le contexte scientifique relatif à l’utilisation d’Avastin.
900. D’ailleurs, s’il est exact que jusqu’à la publication des résultats des premières études de comparaison concernant Avastin et Lucentis, il existait peu de données scientifiques disponibles sur le sujet, comme le souligne l’AFSSAPS dans sa note d’information du 10 septembre 2009 (cf. paragraphes 149 à 152 de la présente décision), il ne saurait en être inféré que l’utilisation d’Avastin « hors AMM » était interdite ou présumée dangereuse. L’AFSSAPS invitait en effet simplement à la prudence dans l’utilisation d’Avastin en intravitréen, sans s’opposer à son utilisation par certains médecins ophtalmologistes.
901. Or, en l’espèce, loin de s’en tenir à un discours évoquant cette situation d’incertitude, la communication de Novartis a été de nature à renforcer les doutes sur l’usage « hors AMM » d’Avastin, en exagérant les risques qui y étaient associés, en comparaison avec l’usage autorisé de son propre produit, Lucentis.
902. Dès lors, l’argument de Novartis sera écarté.
903. Concernant la fin des pratiques, les éléments versés au dossier montrent que, au début de l’année 2014, les équipes commerciales de Novartis continuaient de diffuser un discours insistant sur les risques liés à l’utilisation d’Avastin en ophtalmologie (cf. paragraphe 409 à 415 de la présente décision). Toutefois, comme souligné au paragraphe 759 ci-dessus, la commercialisation d’Eylea pour le traitement de la DMLA, à compter de novembre 2013, a eu pour conséquence de mettre fin à la domination de l’entité collective composée par Genentech, Roche et Novartis sur le marché concerné.
904. Novartis soutient néanmoins que, dans la mesure où la loi Bertrand a interdit l’utilisation « hors AMM » d’Avastin, aucun discours trompeur ne pouvait lui être imputé après décembre 2011.
905. Toutefois, comme indiqué aux paragraphes 641 à 643 de la présente décision, la loi Bertrand n’a pas interdit l’utilisation d’Avastin « hors AMM » en ophtalmologie, et plus particulièrement pour le traitement de la DMLA. Il ressort au surplus des paragraphes 644 à 647 de la présente décision, que même après l’adoption de l’instruction de la DGS de juillet 2012, l’utilisation « hors AMM » d’Avastin en ophtalmologie n’était pas illicite.
906. Dès lors, l’argument de Novartis sera écarté.
907. Par conséquent, il est établi que les pratiques sanctionnées au titre du grief n° 1 ont débuté le 10 mars 2008, pour prendre fin au début du mois de novembre 2013.
Sur les sociétés auteures des pratiques
908. En premier lieu, il ressort des développements qui précèdent que la société Novartis Pharma SAS, qui gère en particulier la franchise Ophtalmologie du groupe pour la France, a élaboré et diffusé le discours faisant l’objet du grief n° 1.
909. Par conséquent, la société Novartis Pharma SAS doit être considérée comme auteure des pratiques reprochées au titre du grief n° 1 ce qui, du reste, n’est pas contesté par les mises en cause.
910. En second lieu, concernant la participation de la société Novartis AG aux pratiques reprochées au titre du grief n° 1, Novartis soutient que cette société ne peut pas être considérée comme co-auteure des pratiques avec sa filiale française, soulignant en particulier que Novartis AG est une société holding sans activité opérationnelle et qu’elle n’apparaît pas dans les pièces utilisées au soutien du grief.
911. Néanmoins, plusieurs pièces du dossier attestent de l’implication du siège du groupe Novartis, désigné par le terme « global », dans l’élaboration du discours examiné au titre du grief n° 1 (cotes 3609, 2567, 3629, 3635 et 2206).
912. Cette situation est cohérente avec la structure du groupe Novartis, organisé en six divisions fonctionnant de façon autonome, parmi lesquelles la division « Pharmaceuticals » qui englobe la franchise mondiale du groupe en ophtalmologie et est responsable de la commercialisation de Lucentis (cotes 387, 391 et 392). D’ailleurs, M. P…, l’auteur de la présentation du 17 mars 2008 « Lucentis : Major Issues & Strategies » était, en 2009, le directeur général de la division « Pharmaceuticals » de Novartis et membre du comité exécutif de Novartis AG(41) (cotes 3609 à 3612).
913. Par conséquent, la société Novartis AG doit être considérée comme co-auteure des pratiques reprochées au titre du grief n° 1. L’argument de Novartis sera donc écarté.
Conclusion générale sur le grief n° 1
914. Il ressort des développements qui précèdent que Novartis a mis en œuvre une pratique de dénigrement d’Avastin, spécialité concurrente de Lucentis, qui a débuté le 10 mars 2008, pour prendre fin début novembre 2013, avec l’arrivée d’Eylea sur le marché.
915. Cette pratique a été de nature à et a eu pour effet de limiter les prescriptions d’Avastin « hors AMM » pour le traitement de la DMLA et, plus généralement, en ophtalmologie. Elle est également susceptible d’avoir eu pour effet le maintien de Lucentis à un prix supra-concurrentiel ou encore la fixation du prix d’Eylea à un niveau artificiellement élevé.
c) Concernant la pratique visée par le grief n° 2
Principes applicables
916. Il ressort de la pratique décisionnelle que des comportements adoptés devant des autorités publiques peuvent, dans certaines circonstances, être examinés au regard du droit des pratiques anticoncurrentielles (décisions n° 05-D-58 du 3 novembre 2005 relative à des pratiques relevées dans le secteur de l’eau potable en Ile-de-France ; n° 16-D-11 du 6 juin 2016 relative à des pratiques mises en œuvre dans le secteur de la diffusion de la télévision par voie hertzienne terrestre et n° 17-D-25 du 20 décembre 2017 relative à des pratiques mises en œuvre dans le secteur des dispositifs transdermiques de fentanyl).
917. Pour apprécier l’existence d’une pratique d’intervention abusive auprès d’une autorité publique, l’Autorité s’attache, non pas à contrôler la légalité de la décision, mais à rechercher si l’entreprise en position dominante s’est immiscée indûment dans le processus décisionnel de cette autorité ou encore si elle a mis en œuvre des pratiques de nature à l’inciter à adopter une décision indue (décisions n° 05-D-58, précitée, points 123 à 125 ; n° 16-D-11, précitée, point 192 et n° 17-D-25, précitée, point 422).
918. Selon la jurisprudence européenne, « la présentation aux autorités publiques d’informations trompeuses, de nature à induire celles-ci en erreur et à permettre, en conséquence, la délivrance d’un droit exclusif auquel l’entreprise n’a pas droit, ou auquel elle a droit pour une période plus limitée, constitue une pratique étrangère à la concurrence par les mérites, qui peut être particulièrement restrictive de la concurrence. Un tel comportement ne correspond pas à la responsabilité particulière incombant à une entreprise en position dominante de ne pas porter atteinte, par un comportement étranger à la concurrence par les mérites, à une concurrence effective et non faussée dans le marché commun » (arrêts du Tribunal du 1er juillet 2010, AstraZeneca, T-321/05, point 355, et de la Cour de justice du 6 décembre 2012, AstraZeneca, C-457/10 P).
919. Ce type de comportement doit être apprécié au vu des circonstances de l’espèce. En effet, « l’appréciation de la nature trompeuse de déclarations fournies aux autorités publiques aux fins de l’obtention indue de droits exclusifs doit être opérée in concreto et est susceptible de varier selon les circonstances propres à chaque affaire ». Plus spécifiquement, « la marge d’appréciation limitée des autorités publiques ou l’absence d’obligation leur incombant de vérifier l’exactitude ou la véracité des informations communiquées peuvent constituer des éléments pertinents devant être pris en considération aux fins de déterminer si la pratique en cause est de nature à aboutir à l’élévation d’obstacles réglementaires à la concurrence » (arrêt du Tribunal AstraZeneca, T-321/05, point 357).
920. De même, comme souligné au paragraphe 775 ci-dessus, la Cour de justice a jugé, dans son arrêt préjudiciel dans l’affaire italienne Roche/Novartis, que des autorités publiques, comme la Commission européenne et l’EMA, étaient susceptibles d’être induites en erreur par une communication d’un laboratoire pharmaceutique sur les risques liés à l’utilisation « hors AMM » d’Avastin (arrêt du 23 janvier 2018, Hoffmann-Laroche, C-179/16, point 92).
921. Par ailleurs, selon les juridictions de l’Union, la réaction des tiers n’exclut pas la responsabilité de l’entreprise en position dominante au titre des articles 102 du TFUE et L. 420-2 du code de commerce. En effet, « la seule circonstance que certaines autorités publiques ne se soient pas laissées abuser et aient décelé les inexactitudes des informations fournies à l’appui des demandes de droits exclusifs, ou que des concurrents aient obtenu, postérieurement à l’octroi irrégulier des droits exclusifs, l’annulation de ceux-ci, ne suffit pas pour considérer que les déclarations trompeuses n’étaient en tout état de cause pas susceptibles d’aboutir. (…) dès lors qu’il est établi qu’un comportement est objectivement de nature à restreindre la concurrence, son caractère abusif ne saurait dépendre des aléas des réactions des tiers » (arrêt du Tribunal AstraZeneca, T-321/05, point 360, voir également arrêt de la Cour de justice, AstraZeneca, C-457/10 P, point 111).
922. Enfin, le caractère délibéré du comportement est susceptible d’être pris en considération. Selon le Tribunal, « si la démonstration du caractère délibéré du comportement de nature à tromper les autorités publiques n’est pas nécessaire aux fins de l’identification d’un abus de position dominante, celui-ci n’en constitue pas moins également un élément pertinent pouvant, le cas échéant, être pris en considération par la Commission » (arrêt du Tribunal AstraZeneca, T-321/05, point 359).
Appréciation en l’espèce
923. Conformément aux principes exposés ci-dessus, il convient d’examiner si la communication mise en œuvre par Novartis et Roche, avec le soutien de Genentech, auprès des pouvoirs publics révèle un comportement de blocage administratif ou revêt un caractère trompeur, et si ce discours a été de nature à influencer la structure du marché.
924. Plus précisément, il ressort des pièces du dossier que les laboratoires membres de l’entité collective ont mis en œuvre des comportements de blocage administratif et sont intervenus de façon inappropriée dans le débat public, par le biais d’un discours alarmiste, voire trompeur, sur les risques liés à l’utilisation d’Avastin en ophtalmologie.
Sur le contenu de la communication de Novartis et de Roche
♦ Concernant les échanges de Roche avec l’AFSSAPS au cours de l’année 2008
925. Comme cela ressort des paragraphes 432 à 437 de la présente décision, l’AFSSAPS a sollicité la collaboration de Roche, dans le cadre d’une réflexion engagée sur l’utilisation d’Avastin « hors AMM » dans le traitement de la DMLA.
926. Toutefois, Roche a adopté à l’égard de l’AFSSAPS une posture de blocage, refusant catégoriquement, et pendant plusieurs mois, de lui fournir les éléments demandés. En effet, il s’est écoulé un an et quatre mois entre la première demande de l’AFSSAPS (courrier du 27 février 2008, cf. paragraphes 436 et 437 de la présente décision) et la date à laquelle Roche a finalement consenti à transmettre les données demandées (le 22 juin 2009, cf. paragraphe 444 de la présente décision).
927. Roche affirme sur ce point que son refus de communiquer les informations en cause était légitime et justifié par une posture de prudence qui ne saurait lui être reprochée. Il soutient également que son discours ne contenait, en tout état de cause, aucun élément trompeur. Enfin, Roche prétend que l’AFSSAPS aurait pu se procurer les échantillons d’Avastin par d’autres moyens.
928. Toutefois, ces arguments ne sauraient prospérer, pour les raisons suivantes.
929. En premier lieu, ce refus ne pouvait être justifié, comme l’indique pourtant Roche dans ses courriers de réponse, par la crainte d’engager sa responsabilité pour avoir encouragé un usage « hors AMM » de son produit. À suivre ce raisonnement, un laboratoire ne pourrait jamais coopérer avec une autorité de santé en vue de l’autorisation administrative d’une nouvelle indication pour un médicament, puisque, par définition, les essais qui seraient alors conduits génèreraient un usage « hors AMM ».
930. D’ailleurs, Roche ne fournit aucun élément concret montrant de quelle façon sa responsabilité aurait pu être engagée en raison de la fourniture de tels échantillons, et ce d’autant moins qu’il était clair, au regard de la demande formulée par l’AFSSAPS, que les produits fournis par Roche n’étaient, en tout état de cause, pas destinés à être injectés à des patients, mais uniquement à réaliser une étude de stabilité de la solution en laboratoire. Ces échantillons étaient en outre requis par une autorité publique ayant pour mission d’examiner la sécurité des médicaments, dans le cadre de ses prérogatives, et le refus de fournir ceux-ci empêchait l’agence de santé de mettre en œuvre les analyses souhaitées.
931. En second lieu, dans ses courriers de réponse aux demandes de l’AFSSAPS, Roche a présenté à l’autorité de santé un discours alarmiste, insistant de manière univoque sur les effets secondaires liés à l’utilisation d’Avastin en ophtalmologie, et a refusé de lui communiquer les données demandées, afin de la dissuader de procéder à l’examen de l’utilisation d’Avastin dans le traitement de la DMLA.
932. Ainsi, sans qu’il soit nécessaire de qualifier de trompeur le discours de Roche dans les courriers des 7 avril et 28 novembre 2008 adressés en réponse aux sollicitations de l’AFSSAPS, il convient de constater que le laboratoire a, par une instrumentalisation des risques associés à l’utilisation d’Avastin en ophtalmologie, cherché à faire obstacle à la volonté de l’AFSSAPS, exprimée publiquement dans un communiqué de presse en février 2007 (cf. paragraphes 432 à 435 de la présente décision), de disposer d’une étude comparative entre Lucentis et Avastin, afin de lui permettre de se prononcer en connaissance de cause en faveur ou en défaveur de l’utilisation d’Avastin pour le traitement de la DMLA.
933. En effet, l’AFSSAPS avait indiqué, à cette occasion, après avoir reconnu l’usage d’Avastin « hors AMM » pour le traitement de la DMLA, qu’elle ne pouvait « pas prendre une position ferme à ce jour contre l’usage d’Avastin », soulignant à cet égard le manque de données lui permettant de conclure (cote 2019).
934. Or, le laboratoire Roche, titulaire de l’AMM d’Avastin et en charge de sa commercialisation en France, était l’interlocuteur incontournable de l’AFSSAPS. S’il est exact que les établissements hospitaliers étaient en mesure de faire remonter à l’autorité de santé des informations sur les risques liés à l’utilisation d’Avastin en ophtalmologie, il n’en demeure pas moins que le laboratoire titulaire de l’AMM, qui est tenu d’assurer un suivi précis de l’utilisation du produit qu’il commercialise au titre de ses obligations de pharmacovigilance, synthétise toutes les remontées d’informations liées à l’utilisation de son médicament et dispose donc, en principe, des données de suivi les plus complètes. C’est d’ailleurs ce qui a conduit Roche à demander, en juin 2011, la modification du RCP d’Avastin, en vue d’y intégrer des informations portant sur des cas d’inflammations de l’œil et d’endophtalmies intervenus après l’utilisation d’Avastin en injection intravitréenne.
935. L’attitude de refus de Roche de communiquer les données demandées à l’AFSSAPS a obligé celle-ci à mobiliser des ressources et du temps pour surmonter les oppositions initiales du laboratoire. En effet, celui-ci aurait pu, et aurait dû – compte tenu de sa responsabilité particulière liée à la détention d’une position dominante collective avec Novartis et Genentech – communiquer dès la première demande de l’AFSSAPS les éléments demandés, tout en assortissant cette communication d’éventuelles réserves quant aux risques dont il souhaitait faire état sur l’utilisation d’Avastin « hors AMM » en ophtalmologie, s’il l’estimait utile et justifié.
936. Pourtant, Roche a opposé deux refus catégoriques à l’AFSSAPS, avant de finalement accepter de transmettre les éléments demandés. Ce faisant, Roche a fait obstacle, de manière injustifiée, à l’exercice de ses missions de surveillance sanitaire. En outre, et en tout état de cause, contrairement à ce que soutient Roche, la réalisation de cette étude de stabilité était indispensable pour la conduite de l’étude GEFAL. En effet, comme l’a indiqué en audition le professeur Gilles Aulagner, promoteur de cette étude : « On avait fait très attention de faire cette étude de stabilité avant le démarrage de GEFAL. On savait que nous devions éviter toute critique de la part des laboratoires » (cote 16575). De même, dans son point d’information du 10 septembre 2009, l’AFSSAPS indiquait : « À ce jour, ces données de stabilité sont donc insuffisantes : c’est la raison pour laquelle la Direction des Laboratoires et des Contrôles de l'Afssaps réalise des études de stabilité de la solution notamment lorsque celle-ci est reconditionnée en seringue ».
937. D’ailleurs, Roche savait que cette étude de stabilité s’inscrivait dans le cadre de la mise en place de l’étude GEFAL, puisque le premier courrier de demande de l’AFSSAPS du 27 février 2008 indiquait expressément que cette étude avait « été décidée par l’Afssaps compte tenu de la mise en place d’un essai clinique ‘GEFAL’ (promoteur : Hospices civils de Lyon) qui compare l’utilisation d’AVASTIN en ophtalmologie (en seringue pré-remplie) à un autre médicament dans le traitement de la DMLA », et soulignait qu’elle s’inscrivait dans le cadre du « protocole clinique » (cote 16398). Roche savait également que les éléments demandés étaient « indispensables », selon les termes de l’autorité de santé dans son courriel de relance du 10 septembre 2008 (cote 16403), pour réaliser les essais de stabilité en question, ce qui ne l’a pas empêché de refuser à nouveau la communication de ces éléments.
938. Il ressort de ce qui précède qu’en refusant, à plusieurs reprises, de répondre favorablement aux demandes de l’AFSSAPS, Roche a retardé, de façon indue, la réalisation de l’étude GEFAL.
♦ Concernant les démarches de Roche et Novartis à l’égard des pouvoirs publics au cours des années 2011, 2012 et 2013
939. Comme cela ressort des paragraphes 445 à 544 de la présente décision, Roche et Novartis ont multiplié les démarches auprès des autorités publiques, tout au long du débat public, politique et administratif ayant conduit à l’adoption de la RTU pour Avastin.
940. Dans ce cadre, les laboratoires ont développé un argumentaire, visant à renforcer les inquiétudes des pouvoirs publics et à faire obstacle aux initiatives de ces derniers, visant à autoriser administrativement l’utilisation d’Avastin pour le traitement de la DMLA.
941. Il convient à cet égard d’examiner le contenu des échanges entre les autorités publiques et Roche, d’une part, et Novartis, d’autre part.
- Les échanges de Roche avec l’AFSSAPS/l’ANSM et d’autres autorités publiques
942. Comme cela ressort des paragraphes 445 à 544 de la présente décision, Roche a échangé à plusieurs reprises, entre 2011 et 2013, avec l’AFSSAPS/l’ANSM et d’autres autorités publiques, au sujet de l’utilisation d’Avastin en ophtalmologie.
943. Dans différents courriers adressés à l’AFSSAPS/l’ANSM, le laboratoire a diffusé à l’autorité de santé un discours alarmiste, insistant de manière univoque sur les effets secondaires liés à l’utilisation d’Avastin en ophtalmologie, en comparaison avec le profil de sécurité établi de Lucentis.
944. Ce faisant, comme lors de ses échanges avec l’AFSSAPS en 2008 (cf. paragraphes 436 à 444 de la présente décision), Roche a, par une instrumentalisation des risques associés à l’utilisation d’Avastin en ophtalmologie, cherché à faire obstacle à la volonté de l’autorité française de santé d’encadrer et de sécuriser les conditions d’usage d’Avastin dans le traitement de la DMLA, compte tenu de l’utilisation constatée en pratique de cette spécialité « hors AMM » par les médecins ophtalmologistes.
945. Dans ses observations en réponse à la notification de griefs et au rapport, Roche met en avant le caractère légitime de ses communications avec l’autorité française de santé, soulignant en particulier que le fait de fournir à cette dernière, tout élément concernant les effets secondaires engendrés par les médicaments qu’un laboratoire commercialise relève de ses obligations de pharmacovigilance. Par ailleurs, en s’appuyant sur l’affaire du Mediator, Roche insiste sur les risques encourus en termes de responsabilité civile et pénale en cas d’utilisation « hors AMM » d’un médicament, justifiant ainsi avoir communiqué sur les risques d’effets indésirables potentiels liés à l’utilisation d’Avastin en ophtalmologie. Enfin, Roche estime que sa mise en cause au titre du grief n° 2 revient à remettre en cause sa liberté commerciale de choisir les indications dans lesquelles il souhaite développer les médicaments qu’il commercialise et son droit de faire valoir ses opinions dans un débat public, protégé par l’article 10 CEDH.
946. Toutefois, ces arguments seront écartés, pour les raisons suivantes.
947. Tout d’abord, les courriers adressés par Roche à l’autorité française de santé s’inscrivent dans un contexte où celle-ci cherchait à prendre position sur l’usage d’Avastin « hors AMM » par les médecins ophtalmologistes, jusqu’alors non encadré, en vue de lui donner, le cas échéant, un cadre réglementaire spécifique.
948. En effet, dans un courrier du 16 mai 2011, l’AFSSAPS indiquait à Roche qu’elle envisageait la mise en place d’un PTT pour Avastin, sous réserve que le laboratoire s’engage à déposer une demande d’AMM pour le traitement de la DMLA, avec une présentation adaptée à cet usage, compte tenu de « l’intérêt du bevacizumab pour la pratique clinique des ophtalmologistes » (cote 6876). De même, dans un courrier du 30 août 2012, l’ANSM demandait à Roche quelles étaient ses intentions concernant l’usage d’Avastin dans les pathologies oculaires, dans un contexte marqué par l’adoption récente de la loi Bertrand et des demandes de RTU, en cours d’instruction, concernant l’emploi d’anti-VEGF pour le traitement de pathologies oculaires, soulignant à cet égard son souhait de « faire bénéficier les patients dans un cadre réglementé équitable et surveillé de nouvelles indications potentielles » (cote 6875).
949. Ainsi, contrairement à ce que soutient Roche, les communications en cause n’intervenaient pas dans le contexte classique de remontée de données de pharmacovigilance concernant le médicament qu’elle commercialise, mais s’intégraient dans le cadre d’initiatives des pouvoirs publics d’encadrer l’utilisation « hors AMM » d’Avastin pour le traitement de la DMLA.
950. Comme le souligne d’ailleurs Roche dans ses observations, les courriers adressés par le laboratoire à l’AFSSAPS/l’ANSM n’ont pas été communiqués de sa propre initiative, mais uniquement en réaction à des sollicitations de l’autorité de santé. Or, loin d’exonérer Roche de sa responsabilité, un tel constat atteste au contraire du comportement de blocage administratif mis en œuvre par Roche, puisque le laboratoire a systématiquement répondu aux demandes de l’AFSSAPS/l’ANSM, par un discours alarmiste sur les risques liés à l’utilisation d’Avastin en ophtalmologie, dont il n’est pas démontré qu’il était justifié par des considérations de santé publique (cf. paragraphes 968 à 974 ci-après).
951. Par ailleurs, outre ses échanges avec l’AFSSAPS/l’ANSM, Roche a également dirigé son discours vers d’autres autorités publiques, comme la ministre de la santé ou la DGS (cf. paragraphes 491 à 494 de la présente décision) au cours de l’été 2012, également saisies dans le cadre du débat public de la question de l’éventuelle autorisation et sécurisation de l’utilisation « hors AMM » d’Avastin, ou, plus généralement, à l’occasion de déclarations publiques en conférences de presse en 2013 (cf. paragraphes 536 et 537 de la présente décision).
952. Or, si un laboratoire pharmaceutique est parfaitement libre de faire valoir, de façon objective et neutre, ses éventuelles préoccupations de santé publique devant les autorités de santé compétentes, il peut en revanche lui être reproché de diffuser auprès d’autorités publiques un discours qui exagère les risques liés à l’utilisation d’un médicament « hors AMM », dans un contexte d’incertitude scientifique, afin de bloquer ou ralentir, indûment, les initiatives visant à encadrer et sécuriser l’usage de ce médicament pour la ou les indication(s) thérapeutique(s) concernée(s). S’il est mis en œuvre par une entité en situation de position dominante, individuelle ou collective, sur un marché, un tel comportement peut être qualifié d’abusif au regard des articles 102 du TFUE et L. 420-2 du code de commerce.
953. Ensuite, le discours diffusé par Roche auprès des autorités sanitaires françaises ne saurait relever de son droit à la liberté d’expression protégé par l’article 10 CEDH, dans la mesure où celle-ci, ainsi que les autres sociétés mises en cause, loin de s’inscrire dans un débat d’intérêt général, ont, par une instrumentalisation des risques associés à l’utilisation d’Avastin en ophtalmologie, uniquement cherché à préserver leurs intérêts commerciaux propres.
954. S’il ne saurait être reproché en soi à Roche – laboratoire commercialisant Avastin en France et titulaire de l’AMM de ce médicament – d’avoir fait part à l’autorité française de santé de l’existence de risques liés à l’utilisation d’Avastin « hors AMM » en ophtalmologie, il n’en demeure pas moins que l’examen de son discours reflète la ligne d’action commune dégagée par l’entité collective, qui n’est pas justifiée par des considérations de santé publique, et constitue une pratique étrangère à la concurrence par les mérites.
955. D’abord, le discours de Roche a été construit et déterminé par anticipation, avant même de recevoir une demande formelle de l’AFSSAPS, comme en témoigne, par exemple, le document du 6 mai 2011, antérieur au premier courrier susvisé de l’AFSSAPS du 16 mai 2011. Dans ce document, Roche expose les « messages clés » du laboratoire, en réaction à la publication des résultats de l’étude CATT. Or, outre la mise en avant des résultats de l’étude CATT « suggérant que le risque d’effets secondaires systémiques seraient moins importants avec injection intra vitréenne de Lucentis comparé à Avastin », ce document contient une présentation de la position de Roche sur l’usage « hors AMM » d’Avastin en ophtalmologie, dans le cadre de laquelle le laboratoire exprime de façon catégorique la supériorité de Lucentis sur Avastin (« Lucentis est le traitement le plus approprié », « Lucentis (…) permet une meilleure pénétration du produit », « Lucentis présente une plus grande affinité » ou encore « Nous restons convaincus que Lucentis est le traitement le plus approprié (gold standard) pour le traitement de la DMLA (forme humide) parce qu’il a été spécifiquement conçu pour une utilisation dans l’œil », cote 6932).
956. Ainsi, si l’expression par un laboratoire pharmaceutique des risques liés à l’utilisation de son propre produit est parfaitement légitime, la pratique, telle que celle exposée en l’espèce, de « promotion » d’un médicament concurrent semble bien éloignée du comportement commercial rationnel d’un opérateur économique.
957. À l’inverse, Roche ne fournit aucun élément interne susceptible de démontrer que sa communication avait pour objectif, ainsi qu’elle le soutient, de minimiser le risque contentieux lié à un possible engagement de sa responsabilité civile ou pénale en cas d’usage « hors AMM » de son médicament.
958. Ensuite, les similarités entre le discours de Roche et celui de Novartis confirment que la communication mise en œuvre par Roche dépassait l’expression de simples préoccupations de santé publique ou, plus généralement, du simple choix du laboratoire de ne pas développer son médicament dans d’autres indications que l’oncologie.
959. En effet, comme exposé aux paragraphes 975 à 1017 ci-après, Novartis a adopté un discours opposant la sécurité d’usage de Lucentis aux incertitudes sur celle d’Avastin. Les deux laboratoires ont ainsi insisté, de la même manière, et selon un argumentaire similaire, sur les risques liés à l’utilisation d’Avastin en ophtalmologie, d’une part, et sur le profil de tolérance bien établi de Lucentis, spécifiquement conçu pour une utilisation dans l’œil, d’autre part, alors pourtant qu’il s’agit d’opérateurs économiques distincts, commercialisant des produits concurrents.
960. Or, ces démarches n’étaient pas motivées par des considérations scientifiques ou sanitaires, ni même par l’affirmation de la liberté commerciale de Roche de ne pas développer son médicament dans des indications en ophtalmologie, mais reflétaient la ligne d’action commune de l’entité collective composée par Roche, Novartis et Genentech, visant à préserver le positionnement respectif d’Avastin et de Lucentis (cf. paragraphes 707 à 725 de la présente décision).
961. Ainsi, contrairement à ce que soutient Roche, il n’est pas reproché au laboratoire d’avoir décidé de ne pas développer Avastin pour des indications en ophtalmologie. En revanche, son discours alarmiste sur les risques liés à l’utilisation d’Avastin en ophtalmologie, associé à la mise en avant de la sécurité établie de Lucentis, relève bien d’une immixtion indue du laboratoire dans les initiatives des pouvoirs publics, qui est susceptible de porter atteinte à une concurrence effective et non faussée sur le marché.
962. Enfin, les critiques du Professeur B…, alors vice-président de la commission d’AMM, sur le projet de courrier de mai 2011 que Roche lui avait soumis avant de l’adresser à l’AFSSAPS, montrent que la présentation des risques dégagés par les études scientifiques comparant Avastin et Lucentis en ophtalmologie a pu être exagérée par le laboratoire (cf. paragraphes 453 à 458 de la présente décision). Si, ainsi que le souligne Roche dans ses observations, ces remarques ont été qualifiées par son auteur de « dures et peut-être un peu injustes », celui-ci a toutefois ajouté « mais pas tant que ça et à l’image des réflexions de beaucoup » (cote 6943). Or, Roche n’a tenu compte de ces observations pour modifier son discours sur les effets indésirables dans un sens plus neutre, ni dans son courrier du 26 mai 2011, ni dans ses courriers postérieurs.
963. De même, dans ses courriers adressés à l’ANSM au cours de l’année 2012, Roche a procédé à une présentation de la décision de l’EMA de modifier le RCP d’Avastin concernant les effets indésirables systémiques, qui ne reflétait pas correctement l’analyse ayant conduit à cette décision. En effet, dans son courrier du 10 septembre 2012, Roche a indiqué : « nous tenons à vous informer que l’Agence Européenne du Médicament a autorisé de nouvelles mises en garde et précautions d’emploi dans le RCP européen d’Avastin. Ces mises en garde décrivent les événements indésirables systémiques et oculaires graves rapportés à la suite d’une utilisation d’Avastin, en injection intra-vitréenne (décision de la Commission Européenne du 30 août 2012) » (cote 16432). Si, comme cela ressort des paragraphes 132 à 139 de la présente décision, l’EMA a effectivement modifié le RCP d’Avastin sur ce point, le CHMP avait, dans son avis de juillet 2012, constaté que « les informations détaillées obtenues des études CATT et IVAN sont rassurantes et il n’y a pas de preuve que le bevacizumab [Avastin] est moins sûr que le ranibizumab [Lucentis] s’agissant des effets systémiques », pour en conclure qu’il était nécessaire d’insérer au RCP d’Avastin un paragraphe sur l’existence d’effets systémiques indésirables liés à l’utilisation des anti-VEGF en intravitréen, sans que cet avertissement concerne spécifiquement Avastin.
964. De la même manière, dans son courrier du 12 novembre 2012, Roche revient, après avoir communiqué les informations sollicitées par l’ANSM, sur les différences moléculaires et les données disponibles de pharmacocinétique de Lucentis et Avastin, et sur la modification susvisée du RCP d’Avastin concernant les effets indésirables systémiques. Ici encore, le laboratoire omet de préciser, comme l’avait souligné le CHMP dans son avis, que le constat ayant abouti à cette modification portait sur l’ensemble des anti-VEGF (dont Lucentis) et non, comme cela est suggéré dans ses courriers, seulement sur Avastin.
965. Ou encore, dans ses déclarations publiques lors d’une conférence de presse le 23 octobre 2013, Roche a insisté de nouveau sur la modification susvisée du RCP d’Avastin, en l’opposant au profil de sécurité bien établi de Lucentis, alors que, après avis du CHMP en janvier 2013, le RCP de Lucentis a été modifié en juillet 2013, afin d’insérer la même mention sur l’existence d’effets indésirables systémiques liés à l’utilisation des anti-VEGF, ce que Roche a omis de préciser et qui modifiait notablement la portée de cette affirmation (cf. paragraphes 140 à 148 de la présente décision).
966. Roche et Genentech soutiennent toutefois que le cadre réglementaire ne permettait pas aux pouvoirs publics d’autoriser administrativement l’usage « hors AMM » d’une spécialité en présence d’une alternative thérapeutique autorisée (Lucentis). Plus spécifiquement, les sociétés mises en cause affirment (i) que l’adoption d’un PTT pour Avastin n’était pas une option envisageable, (ii) que l’instruction de la DGS n’a pas rendu le cadre législatif et réglementaire applicable plus restrictif et (iii) que l’usage d’Avastin pour le traitement de la DLMA ne pouvait pas faire l’objet d’une RTU avant 2015.
967. Ces arguments seront écartés, pour les raisons suivantes.
968. D’une part, les pièces du dossier attestent du fait que l’éventualité de l’adoption d’un PTT pour Avastin en ophtalmologie a été envisagée par l’autorité française de santé. Certes, en février 2007, l’AFSSAPS, qui avait annoncé engager une réflexion sur l’utilisation d’Avastin en ophtalmologie, s’était interrogée sur la mise en place d’un PTT pour cette spécialité. À cette occasion, elle avait indiqué : « Compte-tenu de l'existence de Lucentis, l'Afssaps ne peut pas recommander l'utilisation d'Avastin dans le traitement de la DMLA dans le cadre d'un protocole temporaire de traitement (PTT) mais compte-tenu des remontées (via le groupe de travail), elle ne peut pas prendre une position ferme à ce jour contre l'usage hors-AMM d'Avastin", ajoute-t-on de même source » (cote 2019). Pour autant, cette possibilité ne semblait pas exclue définitivement par l’autorité française de santé, puisque celle-ci a indiqué, quatre ans plus tard, dans un courrier du 16 mai 2011 adressé à Roche, qu’elle envisageait la mise en place d’un PTT pour Avastin compte tenu de « l’intérêt du bevacizumab pour la pratique clinique des ophtalmologistes » (cote 6876).
969. Comme le soutiennent Roche et Genentech, il est vrai que les « Référentiels de bon usage des médicaments de la liste hors-GHS », publiés par l’ANSM en juillet 2012, indiquent que « Les PTT correspondent à des situations hors-AMM acceptables, c’est-à-dire des situations pour lesquelles le rapport bénéfice/risque de la prescription du produit a été évalué comme étant favorable, sur la base des données disponibles, prenant en compte : - qu’il n’existe pas d’alternative thérapeutique ayant l’AMM dans cette situation et présentant une balance bénéfice risque de même niveau, - qu’il n’existe pas de médicament dans le GHS [groupe homogène de séjour] pouvant être prescrit hors-AMM dans cette situation. Cela signifie que l’absence de mise à disposition du traitement pourrait représenter une perte de chance »(42). Toutefois, ces référentiels, établis par les contrats de bon usage des médicaments conclus avec les établissements de santé(43), visent uniquement à organiser les conditions de prise en charge par les régimes obligatoires d’assurance maladie, puisqu’ils permettent de lister des produits utilisés « hors AMM », afin qu’ils soient financés en sus du GHS à l’hôpital (« liste en sus », cf. paragraphe 48 ci-dessus). Au surplus, les décrets n° 2005-1023 et 2008-1121 susvisés ne faisaient pas expressément de l’absence d’alternative thérapeutique une condition d’octroi d’un PTT à une spécialité donnée.
970. En tout état de cause, le grief notifié reproche aux sociétés mises en cause d’avoir fait obstacle à n’importe quelle forme d’« autorisation administrative » de l’usage d’Avastin « hors AMM » dans le traitement de la DMLA, sans viser spécifiquement la délivrance d’un PTT pour Avastin. Or, il existait de nombreux moyens à la disposition des autorités de santé pour se prononcer sur l’usage « hors AMM » d’Avastin dans l’œil. Les différentes prises de position des autorités de santé (points d’information de l’AFSSAPS et recommandations de la HAS) sur le sujet témoignent d’ailleurs de leur capacité à intervenir pour rassurer ou au contraire appeler l’attention sur une pratique médicale donnée.
971. D’autre part, contrairement à ce que soutiennent Roche et Genentech, l’instruction de la DGS de juillet 2012 a contribué à rendre le cadre réglementaire plus restrictif.
972. En effet, premièrement, l’article L. 5121-1 du code de la santé publique n’interdisait pas aux pharmacies à usage intérieur d’hôpitaux de reconditionner Avastin pour un usage en ophtalmologie. Deuxièmement, l’article L. 5121-12-1 du code de la santé publique, dans sa rédaction issue de la loi Bertrand, n’interdisait pas non plus toute utilisation « hors AMM » d’Avastin pour des indications pour lesquelles Lucentis disposait d’une AMM. Troisièmement, si les autorités de santé françaises ont adopté une posture de prudence concernant l’utilisation d’Avastin « hors AMM » en ophtalmologie, elles ne l’ont pour autant jamais interdite, jusqu’à l’adoption de ladite instruction, qui a posé certaines limites à son usage (cf. paragraphes 635 à 651 de la présente décision).
973. Or, il ressort des éléments du dossier que les pratiques en cause ont directement contribué à l’adoption de l’instruction de la DGS de juillet 2012. D’une part, cette instruction paraît à tout le moins à rebours du contexte législatif et scientifique visant à sécuriser et encadrer l’utilisation « hors AMM » d’Avastin en ophtalmologie. En effet, à la même époque, se tenaient les débats parlementaires préalables à l’adoption de la LFSS pour l’année 2013, qui attestent que la proposition de création d’une « RTU économique » visait à répondre spécifiquement à la question des moyens d’assurer « le traitement de la DMLA au meilleur coût pour la société ». De même, à la même époque, l’étude GEFAL, financée par les pouvoirs publics français, visant à étudier les conditions d’efficacité et de sécurité d’utilisation « hors AMM » d’Avastin pour le traitement de la DMLA, était en cours. D’autre part, les éléments du dossier attestent du fait que la DGS a notamment pris en compte les courriers qu’elle a reçus, puisqu’en réponse à une demande d’information des services d’instruction, elle a cité les courriers du 9 mai 2011 et du 3 juillet 2012 reçus de Novartis parmi les documents sur lesquels elle s’est fondée, concernant, plus précisément les cas d’endophtalmies signalés et les risques liés à l’utilisation d’Avastin en ophtalmologie (cote 13458). Or, il convient de rappeler que Novartis n’était pas le titulaire de la spécialité Avastin, objet de ladite instruction.
974. Il ressort de ce qui précède que Roche a, par un discours alarmiste sur les risques liés à l’utilisation d’Avastin pour le traitement de la DMLA, cherché à bloquer ou ralentir, de façon indue, les initiatives des pouvoirs publics qui, malgré le choix fait par ce laboratoire de ne pas développer Avastin pour une indication en ophtalmologie, envisageaient, compte tenu de la pratique des ophtalmologistes, de favoriser et sécuriser son usage pour le traitement de la DMLA.
- Les échanges de Novartis avec les autorités publiques
975. Comme cela ressort des paragraphes 445 à 544 de la présente décision, Novartis a multiplié les démarches vis-à-vis des autorités de santé et des pouvoirs publics, entre 2011 et 2013, dans un contexte de débat public concernant l’utilisation d’Avastin « hors AMM » pour le traitement de la DMLA.
976. Dans ces différents échanges, le laboratoire a diffusé aux autorités publiques un discours alarmiste et trompeur, insistant de manière univoque sur les effets secondaires liés à l’utilisation d’Avastin en ophtalmologie, en opposition avec le profil de sécurité établi de Lucentis.
977. Ce faisant, Novartis a cherché à faire obstacle à la volonté des pouvoirs publics d’encadrer et de sécuriser les conditions d’usage d’Avastin dans le traitement de la DMLA, compte tenu de l’utilisation, constatée en pratique, de cette spécialité « hors AMM » par les médecins ophtalmologistes.
978. Dans ses observations en réponse à la notification de griefs et au rapport, Novartis soutient que, dans un contexte de débat ouvert et relatif à des questions de santé publique, sa communication auprès des autorités publiques relevait de sa liberté d’opinion et d’expression, protégée par l’article 10 CEDH, ainsi que de la défense légitime de ses intérêts. Le laboratoire conteste à cet égard le caractère trompeur de ses communications, compte tenu notamment du contexte dans lequel elles se sont inscrites, de leurs destinataires et de leur objet. Plus particulièrement, Novartis affirme que les éléments contenus dans sa communication sont fondés sur les éléments scientifiques disponibles à cette époque et qu’ils sont précisément référencés afin de permettre aux destinataires compétents d’en apprécier la pertinence.
979. Toutefois, ces arguments seront écartés, pour les raisons suivantes.
980. À titre liminaire, ce n’est pas le principe même de l’intervention du laboratoire auprès des autorités publiques qui est visé dans le grief notifié, mais bien la teneur et les modalités de son discours.
981. En effet, comme souligné au paragraphe
952 de la présente décision, un laboratoire pharmaceutique est parfaitement libre de faire valoir, de façon objective et neutre, ses éventuelles préoccupations de santé publique devant les autorités de santé compétentes. De même, la légitimité des actions de défense de ses propres intérêts (ou « lobbying ») d’un laboratoire pharmaceutique à l’égard autorités publiques ne saurait être remise en cause, dès lors que celles-ci ne méconnaissent par ailleurs aucune législation.
982. Sur ce point, le Conseil de la concurrence a, dans le passé, souligné au sujet d’une pratique mise en œuvre par un syndicat professionnel, que si celle-ci « s’inscrit (...) dans le contexte d’un débat public dans lequel les groupes socio-professionnels font connaître leur point de vue pour défendre les intérêts de leurs membres (…) quand bien même elle conduirait un syndicat professionnel à défendre une position qui ne serait pas la promotion d’une plus forte concurrence, ne peut être qualifiée par elle-même d'anticoncurrentielle » (décision n° 05-D-20 du 13 mai 2005 relative à une saisine de la société le casino du Lac de la Magdeleine, point 27).
983. En revanche, il peut être reproché à un laboratoire de diffuser auprès d’autorités publiques un discours, dont le contenu est trompeur, en ce qu’il exagère les risques liés à l’utilisation d’un médicament « hors AMM », dans un contexte d’incertitude scientifique, afin de bloquer ou ralentir, indûment, les initiatives des pouvoirs publics qui envisagent d’en encadrer et sécuriser l’usage pour la ou les indication(s) thérapeutique(s) concernée(s). S’il est mis en œuvre par une entité en situation de position dominante, individuelle ou collective, sur un marché, un tel comportement peut être qualifié d’abusif au regard des articles 102 du TFUE et L. 420-2 du code de commerce (cf. paragraphes 916 à 922 de la présente décision).
984. Dès lors, c’est au regard du contenu des échanges entre Novartis et les autorités publiques, et du contexte dans lequel ils se sont inscrits, que le comportement du laboratoire doit être appréhendé.
985. Ainsi, concernant d’abord le contexte dans lequel ces échanges ont eu lieu, il ressort des éléments du dossier que Novartis a clairement orienté sa communication vers les autorités chargées de prendre position, dans un contexte de débat public sur l’utilisation d’Avastin en ophtalmologie.
986. À titre d’illustration, dans son courrier du 9 mai 2011, adressé à un grand nombre d’acteurs institutionnels de premier plan dans la définition de la politique de santé (comme le CEPS, l’AFSSAPS et le ministère de la santé, mais également la présidence de la République ou le Premier ministre), Novartis a invité les autorités de santé à prendre position sur les messages portés par les médias tendant à inciter les professionnels de santé à utiliser Avastin « hors AMM », en insistant sur « les risques pour les patients » et les « conséquences importantes en termes de sécurité sanitaire » liés à une telle utilisation (cote 3081).
987. De même, dans la présentation intitulée « PLFSS 2013 - Action plan » d’octobre 2012, Novartis a envisagé plusieurs actions pour lutter contre l’article 45 du PLFSS pour 2013, qui introduisait le principe d’une « RTU économique ». Or, parmi les actions envisagées, figuraient l’envoi de courriers à des personnalités politiques, comme le Premier ministre ou la ministre de la santé (« action 2 ») et des réunions avec les représentants des autorités de santé (« action 3 »), afin de les alerter sur les risques sanitaires et juridiques que l’adoption d’une RTU de cette nature pour Avastin pourraient entraîner, en insistant notamment sur la récente affaire du Mediator (« alert on the major Public Health and sanitary issue that this article and an Avastin RTU would raise (Key messages : risk of safety issues, Mediator like scandal and risk of establishing difference in the access to treatments amongst French population, infringement of Europen pharmaceutical legislation) », cote 1518). Dans la présentation intitulée « LFSS 2013 – Action plan » de février 2013, où Novartis précise les actions à mettre en œuvre contre l’adoption d’une « RTU économique » pour Avastin, en application de la LFSS pour l’année 2013 récemment votée, le laboratoire indique qu’il faut continuer à alerter les principales parties prenantes avec le même type de discours (« continue to alert key stakeholders on issues that this provision raises => major Public Health and sanitary : risk of safety issues, Mediator like scandal and risk of establishing difference in the access to treatments amongst French population, infringement of Europen pharmaceutical legislation », traduction libre : « continuer à alerter les principales parties prenantes sur les problèmes soulevés par cette disposition => importants enjeux de santé public et sanitaire : risques de problèmes de sécurité, scandale comme celui du Mediator et risque d’établir une différence dans l’accès aux traitements au sein de la population française, violation de la législation pharmaceutique européenne », cote 2881).
988. Ainsi, s’il est exact que les autorités de santé ont adopté des positions prudentes concernant l’utilisation « hors AMM » d’Avastin en ophtalmologie (cf. paragraphes 149 à 164 de la présente décision), il n’en demeure pas moins que celles-ci et, de manière plus générale, les autorités publiques, recherchaient une solution juridique permettant de sécuriser et réglementer cet usage, contesté dans le discours de Novartis, mais constaté dans la pratique des médecins ophtalmologistes.
989. Ensuite, les communications de Novartis revêtaient un caractère commercial, s’inscrivant dans le cadre de la ligne d’action commune de l’entité collective, et étaient étrangères à des préoccupations de santé publique.
990. En effet, l’examen du contenu du discours de Novartis, à la lumière du contexte dans lequel il s’est inscrit, montre que celui-ci n’était pas motivé par des considérations de santé publique, mais s’inscrivait dans le cadre d’une manœuvre anticoncurrentielle, visant à empêcher, ou limiter le plus possible, l’utilisation d’Avastin « hors AMM » en ophtalmologie, au profit de son propre produit, Lucentis. Contrairement à ce que soutient Novartis, le discours véhiculé par le laboratoire auprès des autorités publiques revêtait un caractère trompeur.
991. En premier lieu, dans ses échanges avec les autorités publiques, Novartis a fait une présentation sélective, voire erronée, des modifications opérées sur le RCP d’Avastin et de Lucentis.
992. En effet, d’une part, Novartis a, dans certains de ses échanges avec les autorités publiques, indiqué que l’EMA avait procédé à l’interdiction d’utiliser Avastin en intravitréen, ce qui est inexact.
993. Dans son avis de juillet 2012 relatif à la modification du RCP d’Avastin, le CHMP a proposé la modification suivante : « Avastin is not formulated for intravitreal use » (traduction libre : « Avastin n’est pas formulé pour une utilisation intravitréenne », cote 766). Cette modification a été actée, par décision de la Commission européenne, le 30 août 2012 (cf. paragraphe 139 de la présente décision). Ainsi, si un avertissement s’est avéré nécessaire afin de souligner que la forme galénique d’Avastin n’était pas formulée pour une administration intravitréenne, à aucun moment, l’agence de santé européenne n’a interdit l’utilisation d’Avastin en intravitréen.
994. Pourtant, dans un courrier du 9 août 2012 adressé à la DGS, Novartis a indiqué que l’EMA avait modifié le RCP d’Avastin, « pour y ajouter dans sa partie 4.4 ‘Mises en gardes spéciales et précaution d’emploi’, l’interdiction d’utiliser Avastin en intravitréen ainsi que les évènements indésirables oculaires et systémiques qui ont été observés lors de l’utilisation hors AMM d’Avastin en intravitréen » (cote 1529), apportant ce faisant une modification sensible à la portée du changement de RCP. De la même manière, dans un courrier du 5 octobre 2012 adressé au Premier ministre, Novartis a indiqué que l’agence de santé européenne avait interdit l’usage d’Avastin en intravitréen (« circulaire de la DGS du 11 juillet 2012 et précision de l’agence européenne du médicament quant à l’interdiction de l’usage d’Avastin en injection oculaire intravitréenne », cote 1526).
995. Selon Novartis, son affirmation était légitime, puisque la section 4.4 du RCP d’Avastin indiquait que l’utilisation intravitréenne d’Avastin était « non autorisée », en se référant à la rédaction de la section 4 de la notice d’Avastin, qui indique que ce médicament « n’a pas été développé ni fabriqué pour une injection dans l’œil. Il n’est par conséquent pas autorisé pour être utilisé selon cette voie d’administration ». Toutefois, ni cette rédaction, ni celle de la section 4.4. du RCP susvisée, ne signifient que l’usage d’Avastin en injection dans l’œil aurait été interdit par l’EMA, cette mention renvoyant uniquement au fait que l’AMM délivrée à Avastin ne visait pas une utilisation en ophtalmologie. Au surplus, la mention de l’usage « non autorisé » figurait déjà dans la version antérieure du RCP d’Avastin (cote 766).
996. Novartis a donc dénaturé la position de l’agence européenne de santé, cette affirmation, fausse, étant de nature à amplifier la perception, par les destinataires de ce message, des risques liés à l’utilisation « hors AMM » d’Avastin.
997. D’autre part, Novartis a procédé à une présentation de la modification du RCP d’Avastin, suggérant que les effets indésirables oculaires et systémiques détectés concernaient uniquement ce médicament, ce qui est inexact.
998. En effet, pour mémoire, premièrement, le CHMP a considéré qu’il ne convenait pas de faire une distinction entre les différents anti-VEGF, s’agissant des effets systémiques, et, deuxièmement, le RCP de Lucentis contient également un avertissement sur les effets oculaires liés aux injections intra-vitréennes de Lucentis (cf. paragraphes 824 à 832 de la présente décision).
999. Pourtant, dans un courrier du 9 octobre 2012 adressé au président de l’ANSM, un courrier du 16 octobre adressé au responsable adjoint des produits de santé de la CNAMTS ou encore, dans la « Note Lucentis / Avastin » élaborée en préparation des rendez-vous prévus avec le CEPS, la DGS et le cabinet du ministre de la santé, les 11 et 12 octobre 2012, Novartis indiquait que l’EMA « vient, en effet, de modifier le Résumé des Caractéristiques Produit d’Avastin suite à ces différentes études pour y ajouter une mise en garde indiquant qu’Avastin n’est pas formulé pour une utilisation intravitréeenne et préciser les risques oculaires et systémiques en cas d’utilisation intravitréenne » (cotes 15643, 14258, et 14255). Une position similaire est présentée dans la note intitulée « Impact de l’article 57 de la loi LFSS 2013 – Cas Lucentis / Avastin dans le traitement de la DMLA néovasculaire », élaborée en préparation d’une réunion avec le directeur général de l’UNCAM, qui indique notamment « l’EMA a ajouté dans le Résumé des Caractéristiques Produit d’Avastin une mise en garde sur les risques oculaires et systémiques en cas d’utilisation intravitréenne d’Avastin » (cote 14215).
1000.Dès lors, Novartis a, ici encore, fait une présentation des modifications du RCP d’Avastin opérées par l’EMA, dénaturant la portée de la décision de l’EMA, suggérant que ces effets indésirables ne visaient qu’Avastin, alors que le CHMP a estimé qu’il ne convenait pas de faire de distinction entre les différents anti-VEGF, et qu’il a recommandé, en janvier 2013, de porter cette mention au RCP de Lucentis également.
1001.En deuxième lieu, Novartis a effectué une présentation inexacte des prises de position de l’AFSSAPS concernant l’utilisation d’Avastin en ophtalmologie, dans ses deux points d’information du 10 septembre 2009 et du 16 septembre 2011.
1002.En effet, si, à ces deux occasions, l’autorité française de santé a fait référence aux questions de tolérance liées à l’utilisation d’Avastin, en insistant plus particulièrement sur l’absence de forme pharmaceutique adaptée de ce médicament pour un traitement par injection dans l’œil, elle n’a pour autant, à aucun moment, indiqué que cela pouvait « présenter un risque pour la santé publique » (cf. paragraphes 149 à 155 de la présente décision).
1003.Pourtant, dans plusieurs documents destinés aux autorités publiques (le courrier du 3 juillet 2012 adressé à la HAS, cote 15649 ; le courrier du 16 octobre 2012 adressé au responsable adjoint des produits de santé de la CNAMTS, cote 14258 ; ou encore la note intitulée « Note Lucentis / Avastin » élaborée en préparation des rendez-vous prévus avec le CEPS, la DGS et le cabinet du ministre de la santé, les 11 et 12 octobre 2012 (cotes 14162 et 14164), Novartis procède par voie de citation fictive des propos de l’autorité française de santé dans ces deux points d’information, en indiquant que « l’absence de forme pharmaceutique adaptée à l’utilisation en ophtalmologie pour Avastin a été notée par l’ANSM à plusieurs reprises en 2009 et en 2011, comme pouvant ‘présenter un risque pour la santé publique’, notamment un risque infectieux ».
1004.Dès lors, en attribuant à l’autorité française de santé des propos qu’elle n’avait pas véritablement tenus dans les documents visés, Novartis a cherché, ici encore, à renforcer les inquiétudes des autorités publiques, qui sont particulièrement sensibles aux problématiques de santé publique, compte tenu du contexte d’aversion au risque et de judiciarisation des questions de santé décrit ci-dessus (cf. paragraphes 761 à 768 de la présente décision).
1005.En troisième lieu, Novartis a, dans ses échanges avec les autorités publiques, fait une présentation parfois biaisée des résultats des études scientifiques comparant l’efficacité et la sécurité de l’utilisation d’Avastin et de Lucentis en ophtalmologie.
1006.À titre d’illustration, dans son courrier du 9 mai 2011 adressé à un grand nombre d’acteurs institutionnels appartenant ou non au secteur de la santé (comme le CEPS, l’AFSSAPS et le ministère de la santé, mais également la présidence de la République ou le Premier ministre), Novartis a indiqué que « les différents signaux de tolérance liés à l’utilisation hors AMM d’Avastin en injection intra-vitréenne aujourd’hui identifiés dans les trois études précitées [CATT, Curtis et Gower] sont sérieux et ne doivent pas être négligés » (cote 3081). Plus spécifiquement, Novartis a présenté uniquement les résultats des études Curtis et Gower, mentionnant l’existence d’effets secondaires graves plus importants pour un traitement avec Avastin qu’avec Lucentis, et passé sous silence les limites méthodologiques de ces deux études et les conclusions nuancées de leurs auteurs. (cf. paragraphes 98 à 103 de la présente décision).
1007.Novartis soutient toutefois que ce courrier est une réponse très mesurée du laboratoire à l’article, selon elle, agressif et polémique du Figaro, publié le 6 mai 2011, qui soulignait que les deux molécules étaient aussi efficaces, pour une différence de prix très importante (« deux molécules aussi efficaces, l’une à 30 euros, l’autre à 1200 ») et notait, pour ce qui est des effets secondaires « une très légère différence en faveur de Lucentis non significative statistiquement » (cote 3080, cf. paragraphe 460 de la présente décision). Le laboratoire souligne en particulier qu’il a choisi, non pas d’exercer son droit de réponse directement dans le journal accessible au grand public, mais de le cibler vers des destinataires choisis pour leur compétences spécifiques, à même de comprendre des arguments de nature scientifique.
1008.Néanmoins, d’une part, le fait que Novartis ait ainsi agi, atteste au contraire de sa volonté d’intervenir directement auprès des autorités saisies pour étudier et, le cas échéant, décider de l’encadrement éventuel des prescriptions d’Avastin « hors AMM » en ophtalmologie. Or, comme souligné ci-dessus (cf. paragraphe 981 de la présente décision), si le principe des actions de défense de ses intérêts (ou « lobbying ») d’un laboratoire pharmaceutique ne saurait être remise en question, en revanche, un comportement visant, par la diffusion d’un discours dont le contenu est trompeur, en ce qu’il exagère les risques liés à l’utilisation d’un médicament « hors AMM » dans un contexte d’incertitude scientifique, à bloquer ou ralentir indûment les initiatives des pouvoirs publics qui envisagent de favoriser et sécuriser son usage pour le traitement de la DMLA, peut constituer une immixtion indue dans le processus décisionnel des autorités publiques, susceptible d’être contraire aux articles 102 du TFUE et L. 420-2 du code de commerce. D’autre part, force est de constater, en tout état de cause, que la réponse de Novartis à l’article du Figaro a eu une diffusion plus étendue qu’il prétend : ses arguments ont en effet été relayés auprès d’un public plus large, puisqu’ils figurent dans la dépêche APM qui, selon les termes mêmes de Novartis « reprend quasi l’ensemble de nos arguments ! » (cote 3072).
1009.Par ailleurs, dans un courrier du 3 juillet 2012 adressé à la HAS, Novartis a effectué une présentation similaire de ces études, pour en conclure que « les signaux de tolérance liés à l’utilisation hors AMM d’Avastin en injection intravitréenne sont donc sérieux, statistiquement significatifs dans certaines études cliniques » (cote 15649). Or, alors même qu’en réponse à ce courrier, le 31 juillet 2012, la HAS avait, notamment, souligné qu’« il n’y a pas de différences significatives ni en termes d’efficacité ni en termes de tolérance » pour ce qui concerne les effets indésirables systémiques et que, s’agissant des effets indésirables oculaires, « les cas d’endophtalmies, ils sont également décrits avec le Ranibizumab [Lucentis] et ne sont pas significativement plus fréquents dans le groupe Bevacizumab [Avastin] » (cote 3336), Novartis a, dans un courrier du 1er octobre 2012, adressé au directeur général de la DGS, une présentation similaire des effets indésirables graves identifiés dans l’étude CATT pour Avastin, sans tenir compte des nuances apportées par la HAS, et indiqué que les résultats de ces études ont « montré une différence statistiquement significative entre Lucentis et Avastin en défaveur de ce dernier, avec un risque de survenue d’un événement systémique sévère statistiquement supérieur dans le groupe Avastin versus Lucentis » (cote 1528).
1010.De même, dans un courrier du 9 octobre 2012 adressé au président de l’ANSM, en réponse à une demande de l’autorité de santé au sujet de Lucentis, Novartis indique que les études IVAN et CATT « ont mis en évidence une augmentation significative du risque d’événements indésirables graves pour Avastin » (cote 15642), se contentant de présenter les chiffres bruts sans, ici encore, mettre en avant les limites méthodologiques des études. De plus, alors que ce courrier répondait à une demande de l’ANSM concernant Lucentis, le laboratoire a fait le choix de centrer ses développements sur la sécurité d’Avastin, qu’il ne commercialise pourtant pas.
1011.Une position quasi-similaire est présentée par Novartis dans son courrier du 16 octobre 2012 adressé au responsable adjoint des produits de santé de la CNAMTS (cotes 14257 à 14259) ou à l’occasion de nombreuses réunions que Novartis a eues avec les autorités publiques, comme la DGS et le cabinet du ministre de la santé, les 11 et 12 octobre 2012 ou le CEPS, le 12 octobre 2012 (cotes 14161 à 14165).
1012.Dès lors, Novartis a procédé à une présentation parcellaire et décontextualisée des résultats des études scientifiques disponibles.
1013.En dernier lieu, Novartis a diffusé un discours suggérant qu’il pouvait exister un risque d’effets systémiques supplémentaires liés aux propriétés d’Avastin, différentes de celles de Lucentis.
1014.Par exemple, dans la présentation prévue pour le rendez-vous du 12 octobre 2012 avec le CEPS, Novartis insiste sur les « différences structurelles », les « différences pharmacologiques », les « formulations différentes » entre Avastin et Lucentis, pour en conclure que « toutes ces différences impliquent des risques en termes de tolérance locale et systémique » (cote 14619). De la même manière, dans une note intitulée « Note de problématique RTU pour des raisons économiques - projet d’article 45 » adressée le 22 octobre 2012 à des représentants du LEEM afin de les aider à « argumenter », notamment sur les questions de sécurité liées à l’utilisation d’Avastin, dans le contexte des débats publics relatifs à la création d’une « RTU économique » (cote 14253), Novartis souligne les différences entre Avastin et Lucentis (« médicaments différents avec des indications différentes », cote 14255) avant de présenter les conclusions des résultats des études scientifiques, suggérant, ici encore, un lien entre ces différences et le constat desdites études d’une tolérance moindre d’Avastin (cote 14255).
1015.Pourtant, comme souligné au paragraphe 809 de la présente décision, il n’existait pas, au vu des analyses scientifiques disponibles, de lien démontré entre les caractéristiques pharmacologiques respectives des deux produits et l’existence d’effets indésirables systémiques plus importants pour Avastin. D’ailleurs, la HAS a, dans son courrier du 31 juillet 2012, fait part à Novartis de son analyse sur ce point, soulignant que « l’augmentation des effets indésirables mis en évidence dans l’étude CATT ne correspond pas, comme cela est souligné par les auteurs des articles, à un effet pharmacodynamique de l’anti-VEGF » (cote 3336).
1016.Dès lors, la présentation par Novartis des causes potentielles des risques liés à l’utilisation d’Avastin n’a pas été exprimée avec suffisamment de mesure, compte tenu du contexte scientifique dans lequel son discours s’inscrivait.
1017.Il ressort de ce qui précède que Novartis a, par un discours alarmiste et trompeur sur les risques liés à l’utilisation d’Avastin pour le traitement de la DMLA, cherché à bloquer ou ralentir, de façon indue, les initiatives des pouvoirs publics qui envisageaient de favoriser et sécuriser son usage pour le traitement de la DMLA, compte tenu de la pratique des ophtalmologistes.
Sur l’implication de Genentech dans la communication de Novartis et de Roche
1018.Roche et Genentech soutiennent que toute implication individuelle de Genentech doit être écartée. Plus spécifiquement, les sociétés mises en cause allèguent que s’il était légitime que le laboratoire Genentech se tienne informé des évolutions législatives et scientifiques en France concernant les produits que le laboratoire donnait en licence, il ne saurait être considéré qu’il a pris part aux pratiques reprochées, puisqu’il n’est pas présent sur le marché français et qu’il n’a eu aucun contact direct avec les autorités publiques françaises.
1019.Toutefois, plusieurs pièces figurant au dossier attestent du fait que, s’il est vrai que Genentech n’est pas intervenu directement auprès des autorités publiques françaises, il a bel et bien participé à la formulation des réponses à apporter au débat public concernant la commercialisation de ses produits, Avastin et Lucentis, en France. Or, contrairement à ce que soutiennent les mises en cause, ce comportement a excédé le droit de regard légitime d’un donneur de licence sur le développement de son produit dans de nouvelles indications.
1020.En effet, Genentech a transmis à chacun de ses licenciés des informations portant sur les questions de sécurité liées à l’utilisation « hors AMM » d’Avastin en ophtalmologie, afin que celles-ci soient relayées aux médias et aux pouvoirs publics français.
1021.À titre d’illustration, dans un courriel adressé à Roche le 30 juillet 2012, M. M…, dirigeant de Genentech, a indiqué que l’équipe française de Roche devait insister auprès du gouvernement sur le manque de sécurité d’Avastin lorsqu’il est utilisé dans l’œil (« So French team do not need to spend time on checking how the indication can be granted and the label modified, they should rather go back to the government and explain to the above, in particular the lack of safety of Avastin when used in the eye », cote 7148 ; également cote 14689).
1022.Si Roche et Genentech prétendent que M. M… aurait quitté la présidence de Genentech depuis 2010, elles n’apportent pas d’élément probant (pièces contractuelles ou autres) au soutien de cette allégation, alors que les pièces du dossier attestent au contraire que l’intéressé s’exprimait avec une adresse électronique de Genentech et se prévalait de sa fonction de « CEO » de cette société. En effet, d’abord, le curriculum vitae, figurant sur le site internet de Roche, indique qu’en 2009, M. M… était « CEO [Chief Executive Officer]» (président) de Genentech Inc.(44). Ensuite, à l’occasion du courriel litigieux, en 2012, M. M… s’exprimait depuis une adresse électronique de Genentech « M… {CEOs~South San Francisco}" <@gene.com>», cote 7148), qui mentionnait toujours sa fonction de « CEO ». Enfin, la nomination de M. M…, en 2010, en tant que « COO [chief operational officer] » (directeur des opérations) de Roche Pharmaceuticals Division, n’exclut pas qu’il s’exprime au nom de Genentech Inc., puisqu’il est indiqué sur le site du groupe Roche que la division Roche Pharmaceuticals comprend les activités de Genentech(45) et que, dans le cadre de l’accord de fusion entre Roche et Genentech, les locaux de Genentech servent de siège pour l’ensemble des activités pharmaceutiques de Roche aux États-Unis(46). Il paraît au surplus peu crédible qu’un groupe aussi important que Roche ait omis, ainsi que le prétendent les sociétés mises en cause dans leurs observations, de modifier l’adresse électronique et le titre de l’un de ses dirigeants, non pas quelques semaines ou mois après sa nomination, mais deux ans plus tard.
1023.De même, dans un échange de courriels au mois d’août 2012, Genentech a retransmis à Novartis des informations sur la modification du RCP d’Avastin, non encore publiées, obtenues de Roche, en soulignant plus particulièrement l’ajout au RCP d’Avastin d’une mention sur les effets indésirables locaux et systémiques associés à son utilisation en intravitréen : « Please see attached the forthcoming safety update to the European Avastin label which is expected to be finalised in Sept/Oct. Importantly there is mention of both local and systemic side effects associated with unlicensed intravitreal use. We will update the relevant CPO material for rapid distribution once this is finalised and public » (traduction libre : « Veuillez trouver ci-joint la mise à jour à venir du RCP d’Avastin qui devrait être finalisée en septembre/octobre. Il est important de relever la mention de l’existence d’effets indésirables locaux et systémiques associés à l’utilisation ‘hors AMM’ [d’Avastin]. Nous mettrons à jour les documents pour une distribution rapide quand cela sera finalisé et rendu public », cotes 15759 et 15760). Or, rien ne justifiait que Novartis, qui commercialisait un produit concurrent, soit tenu informé par Genentech de cette modification, avant qu’elle soit rendue publique, si ce n’est pour permettre à Novartis de préparer en amont sa communication insistant sur les risques liés à l’utilisation « hors AMM » d’Avastin en ophtalmologie.
1024.Par ailleurs, plusieurs pièces figurant au dossier attestent de l’implication de Genentech dans la définition d’une ligne d’action commune entre les trois laboratoires.
1025.Par exemple, un courriel adressé par Genentech à Roche le 8 mai 2013 atteste de la tenue d’une réunion entre Novartis et Genentech, dont Roche est tenu informé, afin de s’assurer de la diffusion d’un message commun et de l’alignement de leur stratégie de communication, sur la base des « messages clés », dont le contenu a été examiné ci-dessus, insistant notamment sur les risques liés à l’utilisation d’Avastin en ophtalmologie, en comparaison avec la sécurité garantie de Lucentis : « Team is currently making minor revisions to update some details in the key messages but has determined no change in our strategy and overall message is required (…) We're meeting with NVS at 6 pm to discuss and align as much as possible on media strategy and messaging. We anticipate they will be inclined to take an aggressive approach in the media based on potential impact of the GEFAL data on access to Lucentis in France. We anticipate possible media reaction and government pressure on Roche regarding Avastin in France as well » (traduction libre : « L’équipe est actuellement en train de faire des modifications mineures pour mettre à jours des details dans les messages clés mais a décidé qu’aucun changement dans notre stratégie et notre message général n’est requis (…) Nous rencontrons NVS [Novartis] à 18h pour discuter et nous aligner le plus possible sur notre stratégie médias et sur nos messages. Nous anticipons qu’ils auront tendance à adopter une approche agressive dans les médias, autour de l’impact potentiel des données GEFAL sur l’accès de Lucentis en France. Nous anticipons une possible réaction des médias et une pression du gouvernement sur Roche au sujet d’Avastin en France », cote 14736).
1026.De même, comme en attestent les échanges de courriels internes de Genentech du 2 juillet 2012, Genentech a créé une « équipe Avastin » et une « équipe Lucentis », chargées de conseiller les laboratoires sur les informations de sécurité et de tolérance devant être transmises aux autorités de santé françaises : « - Lucentis team to inform Novartis on potential compulsory licensing of Avastin for wAMD in France. - Avastin safety to start potential evaluation of Avastin wAMD safety data transfer to French HA [health authorities] (if requested). - Lucentis team to advise which information on efficacy, publications, etc. regarding Avastin in wAMD should be submitted to French HA [health authorities]. - Lucentis team to advise which information on efficacy, publications, etc. regarding Lucentis in wAMD should be submitted to French HA [health authorities] » (traduction libre : « L’équipe Lucentis va informer Novartis d’une potentielle RTU obligatoire pour Avastin pour la DMLA en France – L’équipe sécurité d’Avastin va commencer l’évaluation potentielle des données de sécurité pour Avastin pour la DMLA à transférer aux autorités françaises de santé – L’équipe Lucentis va conseiller sur les informations sur l’efficacité, les publications etc. sur Lucentis dans la DMLA qu’il conviendra de communiquer aux autorités françaises de santé », cote 15766).
1027.Enfin, comme souligné aux paragraphes 707 à 725 de la présente décision, le rôle particulier de Genentech dans les pratiques en cause se déduit des liens structurels entre les trois laboratoires, et plus particulièrement de la mise en place de comités de gestion et d’un système institutionnalisé d’échanges d’informations. Genentech se trouve en effet au coeur du système contractuel et opérationnel liant les trois laboratoires, qui lui permet de faire la synthèse des enjeux économiques, juridiques et scientifiques pour Avastin et Lucentis, d’intervenir dans la formulation des principes de positionnement respectif des deux médicaments et de déterminer les réponses à apporter au débat public concernant la commercialisation de ses produits en France.
1028.Il ressort de ce qui précède que Genentech a pris part aux pratiques, dans la mesure où il a permis de coordonner le discours de Novartis et de Roche concernant les deux spécialités, s’agissant des données scientifiques. Dans ce contexte, Roche et Novartis ont pu développer un discours très similaire, fondé sur les mêmes éléments techniques, et reprenant les mêmes formulations.
Sur les effets de la communication de Novartis et de Roche
1029.Il convient à ce stade d’examiner les effets, potentiels ou avérés, attachés au discours véhiculé par Roche et Novartis, avec le soutien de Genentech, auprès des autorités publiques, sur le marché français du traitement de la DMLA exsudative et les marchés connexes français des autres pathologies oculaires (OMD, OBVR, OVCR, ou baisse visuelle due à une NVC).
1030.Il ressort des éléments du dossier que les pratiques en cause ont été de nature à avoir, et ont eu, des effets, d’une part, sur les volumes des ventes, et d’autre part, sur les prix des spécialités concernées.
♦ En ce qui concerne l’effet sur les volumes
1031.Compte tenu du contexte dans lequel le discours de Novartis et Roche a été diffusé (cf. paragraphes 761 à 768 de la présente décision), les pratiques des laboratoires commercialisant Lucentis et Avastin ont été de nature à susciter l’inquiétude des autorités publiques sur les possibilités d’utilisation « hors AMM » d’Avastin.
1032.En effet, il convient de tenir compte de l’asymétrie d’information entre les laboratoires pharmaceutiques et la demande, ainsi que de l’aversion au risque des professionnels de santé, en particulier des représentants des autorités de santé, en raison notamment d’une certaine judiciarisation des questions de santé (cf. paragraphes 765 à 768 de la présente décision). Dès lors, toute contestation, fondée ou non, de la sécurité d’utilisation d’un médicament, conduit quasi-inéluctablement à un ralentissement du processus décisionnel des autorités publiques, ce qu’aucun laboratoire pharmaceutique ne peut ignorer.
1033.Plus spécifiquement, en l’espèce, toute appréciation négative à l’égard d’Avastin était de nature à inquiéter les autorités de santé, qui sont attentives aux questions de sécurité liées à l’utilisation des médicaments, compte tenu des enjeux humains qui y sont associés.
1034.En outre, un tel effet a pu être renforcé compte tenu du fait que, non seulement le laboratoire Roche ne s’est pas opposé au discours véhiculé par Novartis – qui jouit d’une réputation particulière dans le domaine de l’ophtalmologie – auprès des autorités de santé, mais il a adopté au contraire une communication similaire à celle de son concurrent, en faveur de l’usage de Lucentis et, de fait, au détriment des ventes de son propre produit, Avastin.
1035.Partant, toute exagération, directe ou indirecte, des risques liés à l’utilisation d’Avastin en ophtalmologie, ne pouvait qu’avoir un effet sensible sur les autorités de santé, soucieuses de protéger les intérêts des patients et que les inciter à recommander aux professionnels de santé de privilégier Lucentis, au détriment d’Avastin.
1036.Ainsi, il ressort des pièces du dossier que les pratiques de Novartis, Roche et Genentech ont eu un impact sur les autorités publiques, et par voie de conséquence, sur l’utilisation « hors AMM » d’Avastin en ophtalmologie et donc, sur la structure de l’offre et de la demande.
1037.En effet, tout d’abord, certains documents internes de Novartis montrent que le discours véhiculé auprès des autorités publiques a eu un effet sur les opinions des responsables publics français. Ainsi, le compte rendu rédigé par le directeur de la « Franchise Ophtalmologie » de Novartis, en vue de son évaluation individuelle pour l’année 2013, fait le lien entre la première communication officielle de Novartis mentionnant les différences de sécurité entre Avastin et Lucentis et le fait que le décret autorisant la publication d’une RTU pour Avastin n’ait toujours pas été publié : « First official Novartis written communication clearly stating Lucentis vs Avastin safety différence, in cooperation with top KOLs. Awareness of these safety issues has really increased in France (cf. Gefal communications, Pr Marananchi audition to senate, CNAM report to Assemblée Nationale, general public media articles,...) which can explain that RTU décret is still not published and that Health Authorities bodies are reorienting their actions towards a Lucentis price discussion more than an Avastin endorsement » (traduction libre : « Première communication écrite officielle de Novartis indiquant les différences entre Lucentis et Avastin en termes de sécurité, en coopération avec les meilleurs KOLs [leaders d’opinion]. L’attention sur ces questions de sécurité a réellement progressé en France (cf. communication Gefal, articles dans les médias…), ce qui peut expliquer que le décret RTU n’est toujours pas publié et que les autorités de santé réorientent leurs actions vers une discussion du prix de Lucentis, plutôt qu’une approbation d’Avastin », cote 45126).
1038.Ensuite, les prises de position officielles des autorités publiques montrent également que ce discours a eu un impact sur celles-ci.
1039.Par exemple, dans son point d’information du 16 septembre 2011, l’AFSSAPS a relayé les réserves de Roche sur la méthodologie de l’étude CATT et sur la tolérance d’Avastin lors d’une utilisation en ophtalmologie (cf. paragraphe 155 de la présente décision). De même, comme souligné au paragraphe 973 de la présente décision, les pratiques en cause ont directement contribué à l’adoption de l’instruction de la DGS de juillet 2012.
1040.Ainsi, le discours des mises en cause a été de nature à décourager les autorités publiques de favoriser un plus large recours « hors AMM » dans le traitement de la DMLA, et plus généralement en ophtalmologie.
1041.Selon les mises en cause, l’utilisation « hors AMM » d’Avastin dans le traitement de la DMLA était marginale et le serait restée, même en l’absence des pratiques. Plus spécifiquement, elles font valoir qu’étant donné l’impossibilité d’obtenir toute forme d’autorisation administrative avant cette date, aucun effet des pratiques ne peut être constaté avant décembre 2014. En particulier, Avastin n’aurait pas pu obtenir de PTT ou de RTU pour son utilisation dans le traitement de la DMLA en raison du contexte réglementaire à l’époque des faits. Ainsi, toujours selon les mises en cause, aucun des outils juridiques à disposition des autorités de santé entre 2008 et la fin de l’année 2014 n’autorisait les pouvoirs publics à favoriser l’usage « hors AMM » d’un médicament en présence d’une alternative thérapeutique autorisée et en l’absence de demande d’AMM. Seule la loi de financement de la sécurité sociale pour 2014 et son décret d’application du 30 décembre 2014 auraient rendue possible l’adoption d’une RTU.
1042.Toutefois, ces pratiques ont été mises en œuvre, comme indiqué aux paragraphes 635 à 647 de la présente décision, alors que le cadre réglementaire n’interdisait pas l’utilisation d’Avastin « hors AMM » pour le traitement des pathologies oculaires, comme le traitement de la DMLA.
1043.De même, s’il est exact qu’il n’existait pas de texte spécifique permettant l’adoption d’une RTU pour Avastin avant l’adoption du décret d’application de la LFSS rectificative pour 2014 (cf. paragraphe 28 de la présente décision), cela ne signifie pas pour autant que les pratiques de Novartis, Roche et Genentech ont été dépourvues d’impact.
1044.Premièrement, le discours de Roche et de Novartis, avec le soutien de Genentech, a contribué au retard dans l’adoption des dispositions législatives nécessaires à l’établissement d’une RTU pour Avastin dans le traitement de la DMLA et, plus généralement, dans l’adoption de dispositions permettant d’encadrer et de sécuriser l’utilisation d’Avastin « hors AMM » en ophtalmologie.
1045.En effet, comme souligné aux paragraphes 925 à 1017 de la présente décision, les comportements poursuivis visaient précisément à retarder l’adoption des dispositions législatives ou réglementaires permettant de sécuriser et d’encadrer l’utilisation d’Avastin « hors AMM » en ophtalmologie. Et, de fait, c’est le constat de l’utilisation d’Avastin « hors AMM » dans le traitement de la DMLA (mais également dans les autres indications oculaires) qui a conduit les pouvoirs publics à modifier la loi, afin de permettre l’adoption d’une RTU lorsqu’il existe déjà une alternative médicamenteuse.
1046.Or, à partir du moment où les pouvoirs publics ont annoncé étudier la possibilité d’une RTU pour Avastin, Roche et Novartis ont multiplié les initiatives et les contacts avec les pouvoirs publics et les autorités de santé pour diffuser leurs messages concernant les risques associés à une telle utilisation (cf. paragraphes 470 à 544 de la présente décision).
1047.En particulier, les 29 et 30 juin 2012, Roche s’inquiétait, auprès du siège du groupe à Bâle, de la volonté des pouvoirs publics d’établir une RTU pour Avastin dans la DMLA : « La Haute autorité de santé (HAS) s'apprête à saisir l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) pour la mise en place d'une recommandation temporaire d'utilisation (RTU) pour Avastin (bévacizumab. Roche) en ophtalmologie (…) Up to now, we didn't receive an official request from HA but we anticipate to receive a request soon and we want the team to be prepared. As usual, we will need to have the position of the team in case the request came officially. We will keep you informed as soon as we receive something new » (traduction libre : « Jusqu’à présent, nous n’avons reçu aucune demande officielle des autorités de santé mais nous pouvons anticiper de recevoir prochainement une demande et nous voulons que l’équipe soit préparée. Comme d’habitude, nous aurons besoin d’avoir la position de l’équipe dans l’hypothèse où la demande arriverait officiellement. Nous vous tiendrons informé dès que nous recevons quelque chose de nouveau », cotes 14706 à 14707). En réponse, Roche a été contacté le 30 juin 2012 par un employé de Genentech, qui a alors transféré aux équipes françaises de Roche les documents contenant la position officielle de Genentech sur le sujet : « Please find attached several documents outlining our position on the off-label use of Avastin in wAMD. Our position has not changed since last year (when the letter was sent by Roche France to the French HA [Health Authorities]) » (traduction libre : « Veuillez trouver ci-joint plusieurs documents décrivant notre position concernant l’usage ‘hors AMM’ d’Avastin dans le traitement de la DMLA. Notre position n’a pas changé depuis l’année dernière [quand la lettre a été envoyée par Roche France aux autorités de santé françaises] », cote 14706). À la suite de cette annonce des pouvoirs publics, Genentech a établi un plan d’action interne, le 2 juillet 2012, distinguant une « équipe Lucentis » et une « équipe Avastin », ayant des missions spécifiques. Ainsi, l’« équipe Avastin » devait notamment commencer à évaluer les informations sur l’usage d’Avastin pour le traitement de la DMLA devant être transmises aux autorités françaises (« Last week, Roche France was made aware that the French HA are evaluating the compulsory licensing of Avastin for wAMD via (a new French law called) RTU […] Avastin Safety to start potential évaluation of Avastin wAMD safety data transfer to French HA (if requested) », traduction libre : « La semaine dernière, Roche France a été avertie que les autorités de santé françaises sont en train d’évaluer la licence obligatoire d’Avastin pour la DMLA via (une nouvelle loi française appelée) RTU (…) La sécurité d’Avastin commencer une potentielle évaluation des données de sécurité d’Avastin pour la DMLA à transferer aux autorités de santé françaises (si demandé) », cotes 15765 à 15766). L’« équipe Lucentis » devait quant à elle fournir des conseils quant au contenu des informations de sécurité et de tolérance, tant de Lucentis que d’Avastin, devant être transmises aux autorités (« Lucentis team to advise which information on efficacy, publications, etc regarding Avastin in wAMD should be submitted to French HA », traduction libre : « L’équipe Lucentis va conseiller sur les informations sur l’efficacité, les publications etc. sur Lucentis dans la DMLA qu’il conviendra de communiquer aux autorités françaises de santé » cotes 15765 à 15766). De même, en août 2012, Roche mentionne en interne qu’« il faudra prévoir pour notre communication avec la DGS et le cabinet de la ministre, de rappeler les conclusions des études CATT, CATT2 et IVAN afin d’argumenter les points de safety et de corriger les mauvaises interprétations […] ce position paper validé par le médical onco et aussi en ligne avec la position de Genentech US sur ce sujet », (cote 7135).
1048.Novartis a également immédiatement réagi à l’annonce dans la presse des travaux des autorités de santé sur l’élaboration d’une RTU pour Avastin et transmis un courrier le 3 juillet 2012 à la HAS, à l’ANSM et au CEPS, réaffirmant sa position quant à l’usage d’Avastin dans le traitement de la DMLA et son interprétation orientée des études de comparaisons (« Aucune spécialité pharmaceutique ne peut donc faire l’objet d’une RTU dans les indications accordées à Lucentis […] Deux études observationnelles de grande ampleur montrent une augmentation statistiquement significative du risque d’accident vasculaire cérébral et de décès sous Avastin par rapport à Lucentis […] les signaux de tolérance liés à l’utilisation hors AMM d’Avastin en injection intravitréenne sont donc sérieux, statistiquement significatifs dans certaines études cliniques », cotes 15646 à 15651).
1049.Ces comportements se sont ensuite poursuivis tout au long du débat parlementaire sur la réforme du mécanisme des RTU proposée dans le PLFSS 2013 (cf. paragraphes 535 à 544 de la présente décision).
1050.De fait, les débats parlementaires sur le PLFSS 2013, qui a introduit la possibilité d’une RTU, mais aussi ceux, deux ans plus tard en juillet 2014, sur le PLFSS rectificatif 2014, qui a conduit à modifier la loi à la suite du rejet par le Conseil d’État, font directement référence, à de multiples reprises, au cas d’Avastin et à la problématique liée à l’énorme différentiel de prix avec Lucentis : « Le problème est que le laboratoire Genentech n’a aucun intérêt stratégique à renoncer à son extraordinaire rente de situation, et donc aucun intérêt non plus à demander cette extension. Ce genre de situation me semble justifier que l’on légifère […] Les pouvoirs publics n’ont pas les moyens d’imposer la mise sur le marché de l’Avastin alors qu’il est beaucoup moins cher. […] C’est la raison pour laquelle nous proposons cet article, qui doit nous permettre de faire face à des situations de ce type […] Je regrette que le laboratoire en question n’ait pas de lui-même, comme je l’y avais invité, proposé une diminution significative du prix de son médicament »(47).
1051.Deuxièmement, le discours des mises en cause a directement contribué à l’interdiction par la DGS de l’utilisation d’Avastin hors AMM en juillet 2012 (cf. paragraphe 973 de la présente décision).
1052.Troisièmement, le discours des laboratoires était de nature à inciter les autorités de santé à reporter une prise de position en faveur de l’utilisation d’Avastin en ophtalmogie ou, à tout le moins, à maintenir une position plus conservatrice que celle qu’elles auraient normalement adoptée, quant à l’utilisation « hors AMM » d’Avastin en ophtalmologie (i.e. points d’information de l’AFSSAPS en 2009 et en 2011, et recommandation de la HAS en 2012).
1053.En particulier, le défaut de communication par Roche des informations demandées par l’AFSSAPS en 2008 a conduit à retarder la mise en place de l’étude GEFAL. Or, comme souligné aux paragraphes 432 à 435 de la présente décision, l’AFSSAPS avait exprimé publiquement son souhait de disposer d’une étude comparant Lucentis et Avastin, afin de lui permettre de se prononcer en faveur ou en défaveur de l’utilisation d’Avastin « hors AMM » dans le traitement de la DMLA.
1054.En ce sens, le protocole de l’étude GEFAL indiquait que « si cette hypothèse se confirmait (efficacité et tolérance au moins équivalentes entre les deux médicaments), cela légitimerait l’utilisation du bevacizumab en pratique courante […] Le laboratoire GENENTECH, détenteur de l'AMM et distributeur des deux médicaments aux Etats-Unis, n'a pas souhaité s'engager dans la promotion d'un essai clinique dans cette indication, de même que les laboratoires responsables de la commercialisation du ranibizumab et du bevacizumab en Europe », (cote 2281).
1055.Enfin, comme souligné au paragraphe 598 de la présente décision, dans la mesure où toutes les pathologies rétiniennes traitées par injections dans l’œil d’anti-VEGF étaient les concernées par le débat sur la comparaison des effets secondaires indésirables entre Avastin et Lucentis, les pratiques mises en œuvre par l’entité collective ont été de nature à affecter les ventes d’Avastin pour le traitement de la DMLA et des autres pathologies oculaires (OMD, OBVR, OBCR ou NVC).
1056.Il ressort de ce qui précède que le discours de Novartis et Roche, avec le soutien de Genentech, a été de nature à décourager les autorités publiques de favoriser un plus large recours à Avastin « hors AMM » dans le traitement de la DMLA, et donc, à limiter les prescriptions d’Avastin « hors AMM » pour cette indication et, plus généralement en ophtalmologie, diminuant ainsi artificiellement la pression concurrentielle exercée sur Lucentis.
♦ En ce qui concerne l’effet sur les prix
1057.Le discours diffusé de Novartis et Roche, avec le soutien de Genentech, a également été de nature à maintenir le prix de Lucentis à un prix supra-concurrentiel, et à conduire à une fixation artificiellement élevée du prix d’Eylea.
1058.En effet, en décourageant les autorités publiques de favoriser un plus large recours à Avastin « hors AMM » dans le traitement de la DMLA, les pratiques des membres de l’entité collective composée par Genentech, Roche et Novartis ont également conduit à reporter la possibilité pour les autorités de santé de pouvoir considérer Avastin comme un comparateur pertinent de Lucentis, ce qui aurait pu entraîner des demandes de baisses significatives du prix de Lucentis et, par voie de conséquence, une fixation du prix d’Eylea à un niveau moins élevé.
1059.Selon les mises en cause, même en l’absence des pratiques, aucune baisse de prix de Lucentis n’aurait pu être négociée par le CEPS.
1060.Toutefois, il a été répondu à ces arguments au stade de l’appréciation des effets du grief n° 1. Ainsi, comme cela a été exposé aux paragraphes 863 à 885 de la présente décision, le CEPS aurait pu procéder à une baisse du prix de Lucentis pendant la période de garantie de prix européen ou, à tout le moins, au moment de la renégociation du prix à l’issue de cette période.
1061.Par ailleurs, les mises en cause soutiennent que la publication des RTU n’occasionne jamais de diminution des prix des traitements que les médicaments obtenant la RTU sont censés concurrencer.
1062.Toutefois, les RTU prises pour exemples par les mises en cause, en ce qu’elles n’auraient pas eu d’effets sur les prix des spécialités concurrentes (i.e., la RTU d’Hemangiol obtenue en février 2016 et la RTU de Lioresal et de Baclofène Zentiva obtenue en mars 2014), ne sont pas directement comparables à la RTU d’Avastin de 2015.
1063.En effet, comme le souligne d’ailleurs l’étude économique de Roche, « Avastin est la seule molécule ayant obtenu une RTU en présence d’une alternative thérapeutique, Lucentis. Pour cette raison, les autres médicaments ayant obtenu une RTU en l’absence de spécialité de même principe actif, de même forme et de même dosage ne disposent en général pas d’une alternative clairement établie », (point 185). Les médicaments cités par les mises en cause ne faisaient donc face à aucune alternative « clairement établie » lorsqu’ils ont obtenu leur RTU.
1064.Par ailleurs, l’écart de prix entre ces produits et leurs alternatives identifiées de façon ad hoc par Roche n’est pas connu, alors que l’écart de coût de traitement entre Lucentis et Avastin correspond à un ratio de 30. Ainsi, du fait d’un différentiel de coût très considérable, l’adoption de la RTU pour Avastin était susceptible d’entraîner une réduction de prix de Lucentis, sans que des effets similaires soient observés pour des RTU concernant des traitements dont le différentiel de coût de traitement avec leurs alternatives est moins important.
1065.Enfin, pour les raisons évoquées au paragraphe 886 de la présente décision, les pratiques ont également pu avoir un effet sur la fixation du prix d’Eylea. En effet, arrivé sur le marché en 2013, son prix aurait pu être fixé à un niveau inférieur, si le prix de Lucentis, utilisé comme comparateur, n’avait pas été maintenu à un niveau supra-concurrentiel, ou si Avastin avait pu, à cette époque, être utilisé comme comparateur direct.
1066.Il ressort de ce qui précède que le discours de Novartis et Roche, avec le soutien de Genentech, est susceptible d’avoir maintenu le prix de Lucentis et permis la fixation de celui d’Eylea, à un niveau artificiellement élevé.
Sur la durée des pratiques
1067.Il convient d’examiner successivement la durée de participation de chacun des membres de l’entité collective.
♦ En ce qui concerne Novartis
1068.Novartis conteste la durée des pratiques retenue à son encontre au titre du grief n° 2. Plus spécifiquement, le laboratoire estime qu’il n’existe pas au dossier d’élément de preuve susceptible d’être retenu à son encontre avant le courrier du 3 juillet 2012 qu’il a adressé à la HAS. S’agissant de la fin de l’infraction, Novartis insiste de nouveau sur le fait qu’Eylea aurait exercé une pression concurrentielle de nature à remettre en cause la ligne d’action commune de l’entité collective avant novembre 2013.
1069.Concernant le début des pratiques, contrairement à ce que soutient Novartis, le premier élément de preuve susceptible d’être retenu à son encontre est le courrier du 9 mai 2011, adressé à un grand nombre d’acteurs institutionnels appartenant ou non au secteur de la santé, dans lequel Novartis a fait une présentation sélective et biaisée des résultats des études scientifiques comparant l’efficacité et la sécurité de l’utilisation d’Avastin et de Lucentis en ophtalmologie (cf. paragraphe 459 de la présente décision).
1070.Concernant la fin des pratiques, comme cela a été souligné au paragraphe 759 de la présente décision, Eylea ne peut être considéré comme ayant constitué un contrepoids sérieux à l’entité collective composée par Genentech, Roche et Novartis, avant novembre 2013.
1071.Les arguments de Novartis seront donc écartés.
1072.Par conséquent, il est établi que les pratiques reprochées à Novartis au titre du grief n° 2 ont débuté le 9 mai 2011 pour prendre fin au début du mois de novembre 2013.
♦ En ce qui concerne Roche
1073.Roche conteste la durée des pratiques qui lui ont été notifiées, compte tenu notamment de l’absence de continuité des pratiques qui lui sont reprochées et de leur nature différente (refus de fournir des échantillons à l’AFSSAPS pour la période comprise entre le 7 avril 2008 et le 22 juin 2009 et communication trompeuse auprès de l’ANSM après mai 2011). Plus spécifiquement, le laboratoire souligne qu’il n’existe au dossier aucun élément susceptible d’être retenu à son encontre entre juin 2009 et mai 2011. Par ailleurs, selon Roche, aucun abus n’est caractérisé avant, au moins, l’été 2012, puisque son courrier du 26 mai 2011 ne présente pas un caractère anticoncurrentiel.
1074.Concernant le début des pratiques, le premier élément de preuve susceptible d’être retenu à l’encontre de Roche est le courrier du 7 avril 2008, par lequel le laboratoire a refusé de donner suite à la demande de l’AFSSAPS (cf. paragraphe 438 de la présente décision).
1075.Contrairement à ce que soutient Roche, les pratiques qui lui sont reprochées ne sont pas de nature différente, puisqu’à l’occasion de ses échanges avec l’AFSSAPS d’abord en 2008, puis avec l’ANSM au cours des années 2011 à 2013, Roche a, par une instrumentalisation des risques associés à l’utilisation d’Avastin en ophtalmologie, cherché à faire obstacle à la volonté de l’autorité française de santé de prendre position sur l’utilisation d’Avastin pour le traitement de la DMLA.
1076.Par ailleurs, s’il est exact que le dossier ne contient pas la preuve d’échanges entre Roche et les autorités publiques entre juin 2009 et mai 2011, il convient de retenir la continuité des pratiques reprochées à Roche, dans la mesure où il n’existe pas de preuve ou d’indice pouvant laisser penser que l’infraction s’est interrompue.
1077.Enfin, contrairement à ce que soutient Roche, dans son courrier du 26 mai 2011, le laboratoire a fait une présentation sélective et biaisée des résultats des études scientifiques comparant l’efficacité et la sécurité de l’utilisation d’Avastin et de Lucentis en ophtalmologie (cf. paragraphe 962 de la présente décision).
1078.Concernant la fin des pratiques, comme cela a été souligné au paragraphe 759 de la présente décision, l’entité collective a pu continuer de mettre en œuvre sa ligne de conduite commune jusqu’à l’entrée d’Eylea sur le marché, en novembre 2013.
1079.Les arguments de Roche seront donc écartés.
1080.Par conséquent, il est établi que les pratiques reprochées à Roche au titre du grief n° 2 ont débuté le 7 avril 2008 pour prendre fin au début du mois de novembre 2013.
♦ En ce qui concerne Genentech
1081.Concernant le début des pratiques, la première manifestation identifiable de l’implication de Genentech dans les pratiques reprochées au titre du grief n° 2 est le message interne du 28 avril 2011 adressé par M. M…, dirigeant de Genentech, aux salariés du groupe Roche, à l’occasion duquel il insiste sur le choix de Genentech de se concentrer sur Lucentis dans le domaine ophtalmologique et de ne pas développer Avastin dans ce domaine.
1082.Roche et Genentech contestent de manière générale l’implication du laboratoire américain dans les pratiques et estiment qu’en tout état de cause, aucun abus n’est susceptible d’être caractérisé avant, au moins, l’été 2012, puisque le message interne du 28 avril 2011 de M. M…, dirigeant de Genentech, sur une application interne au groupe Roche, n’établirait pas un quelconque comportement anticoncurrentiel.
1083.Si, ainsi que le soutiennent Roche et Novartis, ce message n’évoque pas les démarches à entreprendre vis-à-vis des pouvoirs publics, il constitue néanmoins le premier élément attestant du fait que Genentech faisait la synthèse des enjeux économiques, juridique et scientifiques pour les deux molécules et établir une politique commune, permettant par la suite à Roche et Novartis de développer un discours très similaire, fondé sur les mêmes éléments techniques, fournis en grande partie par Genentech (cf. paragraphes 1018 à 1028 de la présente décision).
1084.Concernant la fin des pratiques, comme cela a été souligné au paragraphe 759 de la présente décision, l’entité collective a pu continuer de mettre en œuvre sa ligne de conduite commune jusqu’à l’entrée d’Eylea sur le marché, en novembre 2013.
1085.Les arguments de Roche et Genentech seront donc écartés.
1086.Par conséquent, il est établi que les pratiques reprochées à Genentech au titre du grief n° 2 ont débuté le 28 avril 2011 pour prendre fin au début du mois de novembre 2013.
Sur les sociétés auteures des pratiques
- Concernant les sociétés du groupe Novartis
1087.En premier lieu, il ressort des développements qui précèdent que la société Novartis Pharma SAS, qui gère en particulier la franchise Ophtalmologie du groupe pour la France, élaboré et diffusé le discours faisant l’objet du grief n° 2.
1088.Par conséquent, la société Novartis Pharma SAS doit être considérée comme auteure des pratiques reprochées au titre du grief n° 2 ce qui, du reste, n’est pas contesté par les mises en cause.
1089.En second lieu, concernant la participation de la société Novartis AG aux pratiques reprochées au titre du grief n° 2, plusieurs pièces du dossier attestent en revanche de l’implication du siège du groupe Novartis, désigné par le terme « global », dans les pratiques examinées au titre du grief n° 2 (cotes 15764 et 3041).
1090.Comme souligné au paragraphe 912 ci-dessus, cette situation est d’ailleurs cohérente avec la structure du groupe Novartis, dont la division « Pharmaceuticals » est en charge de la commercialisation de Lucentis (cotes 387, 391 et 392). En outre, lorsque les pouvoirs publics ont commencé à évoquer la possibilité d’une RTU pour Avastin pour le traitement de la DMLA, le siège de Novartis était impliqué directement dans les échanges avec Genentech, au travers en particulier des membres du « Joint Management Committee », (cotes 44924 et 44925).
1091.Par conséquent, la société Novartis AG doit être considérée comme co-auteure des pratiques reprochées au titre du grief n° 2. L’argument de Novartis sera donc écarté.
- Concernant les sociétés du groupe Roche
1092.En premier lieu, il ressort des développements qui précèdent que la société Roche a élaboré et diffusé le discours faisant l’objet du grief n° 2.
1093.Par conséquent, la société Roche doit être considérée comme auteure des pratiques reprochées au titre du grief n° 2 ce qui, du reste, n’est pas contesté par les mises en cause.
1094.En second lieu, comme souligné aux paragraphes 1018 à 1028 de la présente décision, Genentech Inc. a également pris part aux pratiques faisant l’objet du grief n° 2.
1095.Par conséquent, la société Genentech Inc. doit être considérée comme co-auteure des pratiques reprochées au titre du grief n° 2. L’argument des mises en cause sera donc écarté.
Conclusion générale sur le grief n° 2
1096.Il ressort des développements qui précèdent que Roche et Novartis ont, avec l’appui de Genentech, développé et diffusé un discours alarmiste, voire trompeur, auprès des autorités publiques sur les risques liés à l’utilisation d’Avastin pour le traitement de la DMLA, afin de bloquer ou ralentir, de façon indue, les initiatives des pouvoirs publics qui envisageaient de favoriser et sécuriser son usage pour le traitement de la DMLA.
1097.Cette pratique a été de nature à décourager les autorités publiques de favoriser un plus large recours « hors AMM » d’Avastin dans le traitement de la DMLA et, par voie de conséquence, à limiter les prescriptions d’Avastin « hors AMM » pour le traitement de la DMLA et, plus généralement, en ophtalmologie. Elle est également susceptible d’avoir eu pour effet le maintien de Lucentis à un prix supra-concurrentiel ou encore la fixation du prix d’Eylea à un niveau artificiellement élevé.
E. SUR L’IMPUTABILITE DES PRATIQUES
1. LES PRINCIPES APPLICABLES
1098.Il résulte d’une jurisprudence constante que les articles L. 420-2 du code de commerce et 102 TFUE visent les infractions commises par des entreprises.
1099.La notion d’entreprise doit être comprise comme désignant une unité économique, même si, du point de vue juridique, celle-ci est constituée de plusieurs personnes physiques ou morales. C’est cette entité économique qui doit, lorsqu’elle enfreint les règles de concurrence, répondre de cette infraction, conformément au principe de responsabilité personnelle (voir, notamment les arrêts de la Cour de justice du 10 septembre 2009, Akzo Nobel e.a./Commission, C-97/08 P, Rec. p. I-08237, points 55 et 56, et du 20 janvier 2011, General Quimica/Commission, C-90/09 P, Rec. p. I-0001, point 36 ; voir, également l’arrêt de la cour d’appel de Paris du 29 mars 2012, Lacroix Signalisation e.a., p. 18 et 20).
1100.Ainsi, au sein d’un groupe de sociétés, le comportement d’une filiale peut être imputé à la société mère notamment lorsque, bien qu’ayant une personnalité juridique distincte, cette filiale ne détermine pas de façon autonome son comportement sur le marché, mais applique pour l’essentiel les instructions qui lui sont données par la société mère, eu égard en particulier aux liens économiques, organisationnels et juridiques qui unissent ces deux entités juridiques (arrêts Akzo Nobel e.a./Commission, précité, point 58, General Quimica/Commission, point 37, et Lacroix Signalisation e.a., précité, pp. 18 et 19).
1101.Dans le cas particulier où une société mère détient, directement ou indirectement par le biais d’une société interposée, la totalité ou la quasi-totalité du capital de sa filiale auteur d’un comportement infractionnel, il existe une présomption selon laquelle cette société mère exerce une influence déterminante sur le comportement de sa filiale. Dans cette hypothèse, il suffit pour l’autorité de concurrence de rapporter la preuve de cette détention capitalistique pour imputer le comportement de la filiale auteur des pratiques à la société mère. Il est possible à la société mère de renverser cette présomption en apportant des éléments de preuve susceptibles de démontrer que sa filiale détermine de façon autonome sa ligne d’action sur le marché. Si la présomption n’est pas renversée, l’autorité de concurrence sera en mesure de tenir la société mère pour solidairement responsable pour le paiement de la sanction infligée à sa filiale (Akzo Nobel e.a./Commission, précité, points 60 et 61, General Quimica/Commission, points 39 et 40, et Lacroix Signalisation e.a., précité, p. 19-20).
1102.Ainsi que l’a rappelé la cour d’appel de Paris dans son arrêt Lacroix Signalisation e.a., précité, ces règles d’imputabilité, qui découlent de la notion d’entreprise visée aux articles 101 TFUE et 102 TFUE, relèvent des règles matérielles du droit européen de la concurrence. L’interprétation qu’en donnent les juridictions européennes s’impose donc aux autorités nationales de concurrence, lorsqu’elles appliquent le droit européen, ainsi qu’aux juridictions qui les contrôlent (voir également, en ce sens, l’arrêt de la Cour de justice du 4 juin 2009, T-Mobile Netherlands e.a., C-8/08, Rec. p. I-04529, points 49 et 50).
1103.Par ailleurs, comme l’ont rappelé les juridictions tant nationales que de l’Union, cette présomption est compatible avec les principes de responsabilité personnelle et d’individualisation des peines. En effet, lorsqu’une entité économique enfreint les règles de concurrence, il lui incombe, selon le principe de la responsabilité personnelle, de répondre de cette infraction. Ce n’est pas une relation d’instigation relative à l’infraction entre la société mère et sa filiale ni, à plus forte raison, une implication de la première dans ladite infraction, mais le fait qu’elles constituent une seule entreprise au sens des articles 101 TFUE et 102 TFUE de l’Union, qui permet aux autorités de concurrence d’adresser une décision imposant des sanctions à la société mère.
2. APPRECIATION EN L’ESPECE
a) Concernant le groupe Novartis
1104.Les griefs n° 1 et n° 2 ont été notifiés à Novartis Pharma SAS et à Novartis AG, en tant qu’auteures des pratiques, et à Novartis Groupe France SA et Novartis AG, en leurs qualités de sociétés mères.
1105.Comme cela ressort des paragraphes 908 à 913 et 1087 à 1091 de la présente décision, les pratiques sanctionnées au titre des griefs n° 1 et n° 2 ont été mises en œuvre par Novartis Pharma SAS et par Novartis AG.
1106.Pendant toute la période des pratiques, Novartis Pharma SAS était détenue à 100 %, directement ou indirectement, par les sociétés Novartis Groupe France SA et Novartis AG (cf. paragraphe 190 de la présente décision).
1107.En conséquence, il convient d’imputer les griefs n° 1 et n° 2 à Novartis Groupe France SA et Novartis AG, pour les pratiques mises en œuvre par Novartis Pharma SAS et par Novartis AG, ce qui, du reste, n’est pas contesté par les mises en cause.
b) Concernant le groupe Roche
1108.Le grief n° 2 a été notifié à Roche et à Genentech Inc., en tant qu’auteurs des pratiques et à Roche Holding AG, en tant que société mère.
1109.Comme cela ressort des paragraphes 1092 à 1095 de la présente décision, les pratiques sanctionnées au titre du grief n° 2 ont été mises en œuvre par Roche et Genentech Inc.
1110.Pendant toute la période des pratiques, Roche était détenue à 100 %, directement ou indirectement, par la société Roche Holding AG (cf. paragraphe 188 de la présente décision). Par conséquent, il convient d’imputer les pratiques mises en œuvre par Roche à Roche Holding AG en tant que société mère, ce qui, du reste, n’est pas contesté par les mises en cause.
1111.Par ailleurs, Genentech Inc. est détenue à 100 % par Roche Holding AG depuis le 26 mars 2009 (cf. paragraphe 188 de la présente décision). Par conséquent, il convient d’imputer les pratiques mises en œuvre par Genentech à Roche Holding AG en tant que société mère, à compter du 26 mars 2009, ce qui, du reste, n’est pas contesté par les mises en cause.
F. SUR LES SANCTIONS
1112.Les dispositions du I de l’article L. 464-2 du code de commerce et de l’article 5 du règlement (CE) n° 1/2003 du Conseil, du 16 décembre 2002, relatif à la mise en œuvre des règles de concurrence prévues aux articles [101 TFUE] et [102 TFUE] (JO 2003, L1, p. 1) habilitent l’Autorité à imposer des sanctions pécuniaires aux entreprises et aux organismes qui se livrent à des pratiques anticoncurrentielles interdites par les articles L. 420-1 et L. 420-2 du code de commerce, ainsi que par les articles 101 et 102 du TFUE.
1113.Le troisième alinéa du I de l’article L. 464-2 du code de commerce précité prévoit que « les sanctions pécuniaires sont proportionnées à la gravité des faits reprochés, à l’importance du dommage causé à l’économie, à la situation individuelle de l’organisme ou de l’entreprise sanctionnée ou du groupe auquel l’entreprise appartient et à l’éventuelle réitération de pratiques prohibées par le [titre VI du livre IV du code de commerce]. Elles sont déterminées individuellement pour chaque entreprise ou organisme sanctionné et de façon motivée pour chaque sanction ».
1114.Par ailleurs, aux termes du quatrième alinéa du I de l’article L. 464-2 du même code, « le montant maximum de la sanction est, pour une entreprise, de 10 % de montant du chiffre d’affaires mondial hors taxe le plus élevé au cours d’un des exercices clos depuis l’exercice précédant celui au cours duquel les pratiques ont été mises en œuvre. Si les comptes de l’entreprise concernée ont été consolidés ou combinés en vertu des textes applicables à sa forme sociale, le chiffre d’affaires pris en compte est celui figurant dans les comptes consolidés ou combinés de l’entreprise consolidante ou combinante ».
1115.En l’espèce, l’Autorité appréciera ces critères légaux selon les modalités pratiques décrites dans son communiqué du 16 mai 2011 relatif à la méthode de détermination des sanctions pécuniaires (ci-après, le « communiqué sanctions »).
1116.Les sociétés mises en cause ont été mises en mesure de formuler des observations sur les principaux éléments de droit et de fait du dossier susceptibles, selon les services d’instruction, d’influer sur la détermination de la sanction pouvant leur être imposée. La présentation de ces différents éléments par les services d’instruction ne préjuge pas de l’appréciation du collège sur les déterminants de la sanction, qui relève de sa seule compétence.
1. LE GRIEF N° 1
a) Sur la détermination du montant de base des sanctions pécuniaires
1117.Le communiqué sanctions indique que « Le montant de base est déterminé pour chaque entreprise ou organisme en fonction de l’appréciation portée par l’Autorité sur la gravité des faits et sur l’importance du dommage causé à l’économie (…) » (point 22).
1118.La durée des pratiques constituant un facteur pertinent pour apprécier tant la gravité des faits (voir, en ce sens, arrêts de la Cour de cassation du 28 juin 2003, Domo services maintenance, et du 28 juin 2005, Novartis Pharma), que l’importance du dommage causé à l’économie (arrêt de la Cour de cassation du 12 juillet 2011, Lafarge ciments e.a.), elle fera l’objet d’une prise en compte sous ce double angle, selon les modalités décrites dans le communiqué sanctions précité.
Sur la valeur des ventes
Les principes applicables
1119.En application du point 23 du communiqué sanctions, la pratique décisionnelle de l’Autorité retient comme assiette du montant de base pour le calcul de la sanction, la valeur des ventes réalisées par l’entreprise mise en cause pour les biens ou les services qui sont en relation avec l’infraction.
1120.Par ailleurs, selon le point 33 du communiqué sanctions, la valeur des ventes est déterminée par référence au dernier exercice comptable complet de mise en œuvre des pratiques. Toutefois, suivant le point 37 du même communiqué, lorsque ce dernier exercice « ne constitue manifestement pas une référence représentative, l’Autorité retient un exercice qu’elle estime plus approprié ou une moyenne d’exercices, en motivant ce choix ».
1121.Le point 36 du communiqué sanctions permet aussi à l’Autorité, dans les cas où les informations transmises sont incomplètes, de « se déterminer à partir des données dont elle dispose, comme le chiffre d’affaires total, même si ces données sont moins directement en rapport avec l’infraction ou avec les infractions commises et donc moins favorables pour l’intéressé ».
1122.Enfin, selon le point 39 du communiqué sanctions, l’Autorité a la faculté d’adapter l’assiette du montant de base dans les cas où « la référence à la valeur des ventes ou ses modalités de prise en compte aboutirait à un résultat ne reflétant manifestement pas de façon appropriée l’ampleur économique de l’infraction ou le poids relatif de chaque entreprise ou organisme qui y a pris part ».
Appréciation en l’espèce
1123.L’infraction sanctionnée au titre du grief n° 1 – qui a débuté le 10 mars 2008 et s’est achevée début novembre 2013 – concerne les ventes de médicaments pour le traitement de la DMLA exsudative et d’autres pathologies oculaires (OMD, OBVR, OVCR ou baisse visuelle due à une NVC) sur le territoire français.
♦ Sur l’assiette de la sanction pécuniaire de Novartis
1124.Il ressort des éléments du dossier que seule la société Novartis Pharma SAS réalise des ventes en relation avec l’infraction.
1125.Compte tenu de ces circonstances spécifiques propres au groupe Novartis, le prononcé d’une sanction pour chaque société du groupe est inadapté. Or, dans l’hypothèse où un groupe de sociétés, formant une seule et même entreprise au sens du droit de la concurrence, participe à une pratique anticoncurrentielle au travers de plusieurs des personnes morales qui le composent, il est possible de considérer que cet ensemble ne constitue qu’un seul et unique participant à l’entente et non pas plusieurs(48).
1126.En conséquence, la sanction pécuniaire prononcée à l’encontre du groupe Novartis sera assise sur une seule valeur des ventes, correspondant au chiffre d’affaires de la société du groupe commercialisant des produits en relation avec l’infraction, c’est-à-dire celui de Novartis Pharma SAS.
♦ Sur le périmètre de la valeur des ventes
1127.Novartis considère que le périmètre de la valeur des ventes retenue pour déterminer la sanction pécuniaire prononcée au titre du grief n° 1 doit être limité aux seules ventes réalisées à hôpital, dans la mesure où Avastin ne pouvait être prescrit en ville.
1128.Toutefois, comme cela ressort du paragraphe 673 de la présente décision, quand bien même Avastin est un médicament de réserve hospitalière, ce médicament a été utilisé en ville. L’infraction en cause a été de nature à affecter le marché français du traitement de la DMLA exsudative par anti-VEGF et les marchés français des autres indications oculaires (OMD, OBVR, OVCR ou baisse visuelle due à une NVC) traitées par anti-VEGF, combinant la ville et l’hôpital.
1129.En conséquence, il y a lieu de retenir l’ensemble des ventes de Lucentis réalisées par Novartis en France, tant en ville qu’à l’hôpital.
♦ Sur l’exercice de référence
1130.Le dernier exercice complet de participation de Novartis à l’infraction est l’exercice 2012.
♦ Conclusion
1131.Il ressort de ce qui précède que l’assiette sur laquelle sera assise la sanction pécuniaire prononcée au titre du grief n° 1 s’élève à […] euros.
Sur la proportion de la valeur des ventes
Sur la gravité des pratiques
♦ Les principes applicables
1132.Lorsqu’elle apprécie la gravité d’une infraction, l’Autorité tient notamment compte du secteur concerné, de la nature de l’infraction et de leurs caractéristiques objectives, telles que leur degré de sophistication (voir point 26 du communiqué sanctions).
1133.Plus spécifiquement, l’Autorité considère, de façon constante, que les pratiques intervenant « dans le secteur de la santé, dans lequel la concurrence est déjà réduite en raison de l’existence d’une réglementation destinée à assurer le meilleur service pour la population tout en préservant les équilibres budgétaires du système d’assurance maladie » sont, de manière générale, particulièrement graves. L’Autorité a ainsi considéré, s’agissant de l’accès à différentes catégories de médicaments, que les pratiques qui visent à entraver l’entrée sur le marché et le développement de médicaments génériques sont d’autant plus graves qu’elles contreviennent à l’objectif affiché des pouvoirs publics de réduire les dépenses de santé, dans un contexte budgétaire fortement contraint (voir la décision n° 13-D-11, précitée, confirmée par les arrêts de la cour d’appel de Paris du 18 décembre 2014 et de la Cour de cassation du 18 octobre 2016, ainsi que la décision n° 13-D-21 précitée, confirmée par les arrêts de la cour d’appel de Paris du 26 mars 2015 et de la Cour de cassation du 11 janvier 2017).
♦ Appréciation en l’espèce
1134.Il ressort des développements qui précèdent que Novartis a mis en œuvre une politique de communication dénigrante, en exagérant de manière injustifiée les risques liés à l’utilisation d’Avastin « hors AMM » pour le traitement de la DMLA, et plus généralement en ophtalmologie, en comparaison avec la sécurité et la tolérance de Lucentis pour un même usage.
1135.Les pratiques en cause sont particulièrement graves, car elles sont intervenues dans le secteur de la santé, où la concurrence est limitée, et plus spécifiquement, dans un contexte de débat public sur le prix extrêmement élevé de Lucentis et son impact sur les finances sociales, pour lesquelles le remboursement à 100 % de Lucentis constituait un poste de dépense significatif, alors qu’il existait un médicament, Avastin, nettement moins cher, susceptible d’être utilisé en ophtalmologie.
1136.Or, par sa politique de communication dénigrante, Novartis a cherché à faire échec aux initiatives des médecins ophtalmologistes qui, dans le cadre de leur liberté de prescription, décidaient de prescrire Avastin « hors AMM » en ophtalmologie, dans le cadre de leur appréciation médicale, en tenant compte de l’intérêt du patient mais aussi de considérations économiques (impact sur le budget de l’hôpital ou sur les dépenses de l’assurance maladie). À titre d’illustration, le chef de service de pharmacie clinique du groupe hospitalier Hôpitaux universitaires Paris Centre indiquait, dans un courrier de janvier 2012 adressé à la DGS avoir « fait le choix d’utiliser Avastin (bevacizumab) en dehors de l’AMM plutôt que Lucentis (ranibizumab) selon les modalités de l’AMM, non seulement pour des raisons économiques (Lucentis est en effet 40 fois plus coûteux que notre préparation et cette attitude nous a permis d’économiser 3,5 millions d’euros en 5 ans) mais en fondant notre analyse sur diverses publications scientifiques » (soulignement ajouté, cote 7105).
1137.Les pratiques en cause sont d’autant plus graves qu’elles présentent un fort degré de structuration et de sophistication. En effet, d’une part, le discours véhiculé par Novartis a évolué en fonction du contexte scientifique et réglementaire, le laboratoire procédant à une mise à jour fréquente de son discours. Loin d’être une prise de position unique à un moment donné, le discours a été renouvelé, par le laboratoire, sur plusieurs années : la constance dans la répétition de ce discours révèle l’importance pour Novartis d’introduire le doute dans l’esprit de ses interlocuteurs, et de modifier leurs comportements pour réduire à néant l’utilisation d’Avastin en ophtalmologie. D’autre part, le discours du laboratoire s’est décliné, de manière structurée, sous plusieurs formes (« messages clés », « questions / réponses » ou encore « Elevator speech ») et a été diffusé selon différentes modalités, afin de toucher le plus grand nombre de destinataires possibles (médecins, associations de patients, grand public). Plus particulièrement, Novartis a adopté une stratégie de communication différenciée selon les destinataires, en insistant spécifiquement sur les KOL, ou encore, en ciblant ses actions sur les centres d’injection connus pour utiliser Avastin en ophtalmologie (cf. paragraphes 788 à 791 de la présente décision). Le discours de Novartis a ainsi directement interféré avec l’exercice par les médecins de leur activité, pour les dissuader de favoriser le recours à Avastin en ophtalmologie (voir par exemple, dans le cas du professeur Y... : « Key lever : Keep Y… busy », traduction libre : « Un levier essentiel : maintenir Y… occupé », cote 13996), alors que ceux-ci ne disposaient pas du temps pour décrypter le message reçu du laboratoire, en allant le comparer le texte intégral des études ou les mentions du RCP.
1138.Novartis fait valoir, pour contester la gravité des pratiques, que son discours auprès des professionnels de santé, des associations de patients ou des autorités de santé, ne s’inscrivait pas dans une quelconque stratégie anticoncurrentielle. Le laboratoire soutient par ailleurs que ses efforts de mise à jour de ses supports de communication reflètent au contraire son souci de demeurer exact et à jour dans sa communication. Enfin, Novartis affirme que les destinataires de son discours étaient parfaitement en mesure de comprendre les problématiques du débat scientifique en cours concernant l’utilisation d’Avastin « hors AMM » en ophtalmologie et qu’en conséquence, aucun discours n’était susceptible de les tromper.
1139.Toutefois, d’une part, il ressort des développements qui précèdent que le discours de Novartis n’était pas motivé par des considérations de santé publique, mais visait à empêcher l’utilisation d’Avastin « hors AMM » en ophtalmologie, au profit de son propre produit, Lucentis (cf. paragraphe 799 de la présente décision).
1140.D’autre part, il n’est pas contesté que les médecins destinataires du discours de Novartis disposent de connaissances scientifiques leur permettant notamment de se faire une opinion sur les résultats des études scientifiques concernant l’utilisation « hors AMM » d’Avastin en ophtalmologie. Toutefois, compte tenu du contexte d’aversion au risque des professionnels de santé et de la judiciarisation croissante des questions de santé, toute contestation, fondée ou non, de la sécurité d’utilisation d’un médicament, associée à un discours insistant sur la responsabilité susceptible d’être engagée par les médecins le prescrivant, conduit quasi-inéluctablement à freiner son utilisation, ce qu’aucun laboratoire pharmaceutique ne peut ignorer. En outre, la méthode adoptée par Novartis visant à s’adjoindre le concours de KOL visait précisément à accroître la crédibilité scientifique de son discours sur les médecins.
1141.Dès lors, les arguments de Novartis seront écartés.
1142.Il ressort de ce qui précède que les pratiques sanctionnées au titre du grief n° 1 doivent être considérées comme très graves.
Sur le dommage causé à l’économie
♦ Les principes applicables
1143.L’Autorité doit procéder à une appréciation de l’existence et de l‘importance du dommage à l’économie en se fondant sur une analyse aussi complète que possible des éléments du dossier et en recherchant les différents aspects de la perturbation générale du fonctionnement normal de l’économie engendrée par les pratiques en cause (arrêt de la cour d’appel de Paris du 30 juin 2011, Orange France, n° 2010/12049, p. 5, confirmé sur pourvoi par arrêt de la Cour de cassation du 30 mai 2012, précité, et du 26 janvier 2012, Beauté prestige international, précité, p. 89). Dans un arrêt du 26 janvier 2010, Adecco France, la cour d’appel a précisé s’agissant du périmètre de cette perturbation générale que « l'appréciation de l'importance du dommage à l'économie (…), qui ne se limite pas, par principe, à la seule atteinte au surplus économique des consommateurs, doit porter sur la perte du surplus subie par l'ensemble des opérateurs du marché, entreprises concurrentes, offreurs ou demandeurs ».
1144.Les effets tant avérés que potentiels de la pratique peuvent être pris en considération à ce titre (voir, en ce sens, arrêt de la Cour de cassation du 28 juin 2005, Novartis Pharma, n° 04-13910). Par ailleurs, si l’existence du dommage à l’économie ne peut être présumée, et s’il doit s’apprécier de manière objective et globale en prenant en compte l’ensemble des éléments pertinents de l’espèce, pour autant, l’Autorité n’a pas à quantifier le dommage à l’économie (arrêt de la Cour de cassation du 7 avril 2010, Orange France e.a., précité).
1145.En se fondant sur une jurisprudence établie, l’Autorité tient notamment compte, pour apprécier le dommage, de l’ampleur de l’infraction, telle que caractérisée entre autres par sa couverture géographique ou par la part de marché cumulée des parties dans le secteur concerné, de sa durée, de ses conséquences conjoncturelles ou structurelles, ainsi que des caractéristiques économiques pertinentes du secteur concerné (voir, par exemple, l’arrêt de la cour d’appel du 30 juin 2011, Orange France, précité).
♦ Appréciation en l’espèce
- Sur l’ampleur des pratiques
1146.Les pratiques visées par le premier grief ont été mises en œuvre sur l’ensemble du territoire national, par une entreprise disposant d’une part de marché très importante. En effet, dès sa mise sur le marché, Lucentis a atteint 77 % des ventes pour le traitement de la DMLA, puis a vu sa part de marché progresser. Par ailleurs, comme souligné aux paragraphes 178 à 180 de la présente décision, l’instruction de la DGS de juillet 2012 a conduit la plupart des ophtalmologistes utilisant précédemment Avastin « hors AMM » à cesser une telle utilisation, laissant Lucentis en position de quasi-monopole pour le traitement de la DMLA, jusqu’à l’arrivée d’Eylea. Les pratiques ont précisément visé à diminuer la part de marché d’Avastin et l’influence que ses prescriptions pouvaient avoir à la fois sur les ventes de Lucentis et sur la position du CEPS sur le niveau de prix de Lucentis.
1147.Par ailleurs, au cours de la période des pratiques, le principal concurrent de Lucentis était Avastin, distribué en France par le laboratoire Roche, avec lequel Novartis détenait collectivement, avec Genentech, une position dominante. L’ampleur des pratiques a ainsi été renforcée en ce que ni Roche, ni Genentech, du fait des liens structurels et contractuels qui les unissaient à Novartis, n’avaient d’incitations à s’opposer aux pratiques de Novartis et, au contraire, avaient défini un plan d’action commun visant à dissuader l’extension de l’usage d’Avastin pour traiter la DMLA et les autres affections oculaires.
1148.Ainsi, jusqu’à l’entrée d’Eylea sur le marché concerné, aucun acteur extérieur à l’entité collective n’était susceptible de remettre en cause le résultat attendu des pratiques mises en œuvre par Novartis.
1149.L’ampleur de la pratique s’apprécie également au travers de ses caractéristiques. Or, Novartis a diffusé un discours exagérant de manière injustifiée les risques liés à l’utilisation « hors AMM » d’Avastin dans le traitement de la DMLA, et plus généralement en ophtalmologie, auprès de l’ensemble des parties susceptibles d’influer sur les prescriptions de Lucentis et d’Avastin : professionnels de santé, et notamment KOL, associations de patients et autorités de santé.
1150.Par ailleurs, contrairement à ce qu’avancent Novartis et Roche, toutes les indications ophtalmologiques d’Avastin ont été affectées par la pratique. En effet, les anti-VEGF – Avastin, Lucentis et Eylea – permettent de traiter à la fois la DMLA et d’autres pathologies oculaires (comme l’OMD, l’OBVR ou OVCR ou encore la baisse visuelle due à une NVC). En outre, ces mêmes anti-VEGF sont le traitement de première intention pour ces pathologies (paragraphe 60 de la présente décision). Comme cela ressort des paragraphes 78 et 86 de la présente décision, Lucentis et Eylea ont d’ailleurs obtenu progressivement des AMM pour ces différentes indications. Or, avant que Lucentis obtienne une AMM pour les pathologies autres que la DMLA, Avastin était également largement utilisé pour le traitement de ces autres indications, comme l’ont indiqué plusieurs médecins auditionnés pendant l’instruction (paragraphes 177 et 654 de la présente décision et cotes 7090, 7095, 7096, 13585 et 13589). Ainsi, la structure des marchés de ces autres indications est sensiblement identique à celle du marché correspondant au traitement de la DMLA.
1151.Les documents internes de Novartis montrent d’ailleurs qu’il s’inquiétait également de l’utilisation particulièrement importante d’Avastin pour ces autres indications. Selon ces documents, sa stratégie consistait précisément à bloquer l’utilisation d’Avastin dans ces autres indications, dans l’attente d’une AMM de Lucentis : ainsi, pour le CHU de Besançon et le CH de Vesoul, l’objectif de Novartis était de « s’opposer au référencement d’Avastin en ophtalmo pour indications OMD et OVR » et de « transférer les unités d’Avastin utilisées dans ces indications en unités de Lucentis » (juin 2012, cotes 15696 à 15700). De fait, toutes les pathologies rétiniennes traitées par injections dans l’œil d’anti-VEGF sont concernées par le débat sur la comparaison des effets secondaires indésirables entre Avastin et Lucentis : la question de la tolérance et des effets indésirables est indifférente à la pathologie exacte pour laquelle ces produits sont utilisés (DMLA ou autres). Les volumes de ventes d’Avastin pour ces autres indications ont donc pu être affectés de la même manière par les comportements des mises en cause.
1152.En tout état de cause, la DMLA reste de loin la principale indication des anti-VEGF. En effet, avant juin 2011, la DMLA est la seule indication traitée avec Lucentis. En 2018, un rapport de la HAS sur l’utilisation de Lucentis montre que la DMLA représente près de 75% des patients traités par ce médicament, contre près de 16 % pour l’OMD(49).
1153.Enfin, l’ampleur des pratiques mises en œuvre par Novartis ressort également de la durée de leur mise en œuvre et de l’importance du marché en cause. En effet, le discours reproché à Novartis a été diffusé pendant près de six ans, à l’ensemble des parties prenantes du secteur français de la santé.
- Sur les caractéristiques des marchés affectés
1154.Les caractéristiques économiques du secteur en cause susceptibles d’influer sur l’importance du dommage à l’économie sont le différentiel de coût des traitements de la DMLA, les barrières à l’entrée, les modalités de négociation du prix et l’importance des questions de sécurité des médicaments.
1155.En effet, le dommage à l’économie causé par les pratiques est amplifié par le différentiel de coût extraordinairement important existant entre Avastin et Lucentis. Chaque flacon d’Avastin pouvait être utilisé pour effectuer plusieurs injections (cote 7104), ramenant le coût unitaire par injection à environ 30 euros (cote 2430), alors que le prix facial de Lucentis a évolué entre 1 161 euros et 647 euros entre 2007 et fin 2015 (soit un différentiel d’un facteur 20 au minimum, voire de 40 au début de la période). Si le prix de Lucentis était insusceptible de diminuer jusqu’au niveau du coût de traitement d’Avastin et si Lucentis pouvait présenter des avantages par rapport à Avastin, à la fois en raison de son AMM et des modalités d’utilisation d’Avastin (e.g., nécessité d’une hotte pour le reconditionnement notamment), un tel différentiel de coûts implique qu’une perte de part de marché, même minime, d’Avastin a des répercussions importantes sur les dépenses de santé. À titre illustratif, un point de pourcentage de parts de marché perdu par Avastin au profit de Lucentis représente un surcoût d’environ 230 000 euros par mois(50). Ce différentiel de coût explique également qu’une fois l’équivalence thérapeutique entre ces deux traitements établie, le CEPS aurait pu chercher à s’appuyer sur l’utilisation par les ophtalmologistes de ce produit pour renégocier à la baisse le prix de Lucentis. À titre illustratif, une baisse de 1% du prix de Lucentis aurait généré en moyenne une économie d’environ 240 000 euros par mois(51).
1156.Le secteur présente en outre d’importantes barrières à l’entrée, résultant notamment des coûts de développement considérables d’un nouveau médicament. Toute entrée d’un produit concurrent exige au minimum plusieurs années de développement et d’études cliniques, et des investissements importants. Par conséquent, le fait qu’un laboratoire parvienne à réduire la pression concurrentielle exercée par un autre traitement est de nature à décourager l’entrée à court terme d’autres traitements concurrents. Au cas d’espèce, la seule alternative à Avastin pour concurrencer Lucentis était le traitement Eylea, qui n’a cependant pu entrer sur le marché concerné qu’à compter de novembre 2013.
1157.Par ailleurs, s’agissant des modalités de négociation du prix des médicaments, celui-ci résulte d’une négociation entre, d’une part, le laboratoire titulaire et, d’autre part, le CEPS. Or, la pratique de Novartis a exagéré, dans un contexte d’incertitude scientifique, le doute sur la valeur thérapeutique et la sécurité d’Avastin en ophtalmologie, qui ne pouvait être levé que par un processus scientifique et administratif long et coûteux, et qui a réduit les incitations des ophtalmologistes à utiliser cette spécialité « hors AMM ». Par voie de conséquence, ces pratiques ont été de nature à empêcher le CEPS de considérer qu’Avastin pouvait être un comparateur pertinent de Lucentis.
1158.Enfin, la demande de médicaments exprimée par les praticiens est généralement extrêmement sensible aux informations disponibles quant à la sécurité de ces médicaments. Des pratiques tendant à remettre en cause la tolérance et l’innocuité d’un médicament sont particulièrement à même d’influencer les comportements des prescripteurs. Ainsi, en dépit du différentiel de coût très important entre Avastin et Lucentis, le discours développé par Novartis, centré sur les questions de sécurité, n’était pas susceptible d’être réévalué à l’aune de considérations économiques et pouvait donc entraîner une forte diminution de la part de marché d’Avastin, et partant, une forte hausse des coûts des traitements pour l’Assurance maladie et les autres organismes d’assurance.
- Sur les conséquences structurelles et conjoncturelles
1159.En premier lieu, comme évoqué aux paragraphes 839 à 856 de la présente décision, les pratiques mises en œuvre par Novartis ont eu pour conséquence de limiter l’usage, par les médecins ophtalmologistes, d’Avastin « hors AMM » en lieu et place de Lucentis.
1160.Ainsi, au début de la commercialisation de Lucentis, les pièces du dossier montrent qu’Avastin était bien utilisé en dehors des usages ayant justifié l’AMM par une certaine proportion des praticiens français, et ce en dépit de freins indépendants des pratiques et intrinsèques à son utilisation « hors AMM » (i.e., nécessité d’un équipement adéquat pour le reconditionnement stérile d’Avastin, obligation d’information des patients par les prescripteurs, péremption des seringues d’Avastin, recours d’Avastin devant être considéré comme indispensable par le médecin prescripteur).
1161.Dans ce contexte, Novartis a cherché, tout d’abord, à limiter la part de marché d’Avastin en dessous de 10 % pour le traitement de la DMLA (cf. paragraphe 846 de la présente décision). Les pratiques de Novartis ont par ailleurs eu un impact sur les opinions des ophtalmologistes et sur les décisions prises par les professionnels de santé concernant l’usage d’Avastin « hors AMM » (cf. paragraphe 848 de la présente décision). Enfin, les résultats de sondage utilisés en interne par Novartis confirment la diminution des parts de marché d’Avastin dans le traitement de la DMLA (cf. paragraphe 849 de la présente décision). Dès lors, par son discours, Novartis a fait en sorte que les transferts de prescription au profit d’Avastin n’interviennent pas.
1162.Pris ensemble, ces éléments attestent bien d’une diminution significative de la part de marché d’Avastin, en raison des pratiques litigieuses, et ce dès le début de l’année 2008 et jusqu’en juin 2012, l’instruction de la DGS de juillet 2012 conduisant alors à dissuader quasi-totalement l’utilisation d’Avastin contre les troubles ophtalmologiques. Or, comme indiqué supra, le différentiel de prix entre Avastin et Lucentis et les volumes de ventes concernés font qu’une telle diminution de la part de marché d’Avastin, et ce pendant plusieurs années, a eu une incidence négative sur l’évolution des dépenses de santé, même en tenant compte du fait qu’une progression des ventes de Lucentis a permis de négocier des baisses de prix de ce traitement.
1163.En second lieu, comme développé supra (cf. paragraphes 857 à 888 de la présente décision), le discours diffusé par Novartis a également pu avoir pour effet indirect de maintenir le prix de Lucentis à un prix supra-concurrentiel.
1164.En effet, une utilisation plus significative d’Avastin « hors AMM » en opthalmologie aurait pu modifier l’appréciation portée par le CEPS sur la valeur thérapeutique d’Avastin par rapport à Lucentis et, par conséquent, le résultat des négociations sur le niveau de prix du Lucentis. À cet égard, il ressort des pièces internes de Novartis figurant au dossier que le discours mis en œuvre par Novartis vis-à-vis des praticiens visait à préserver le niveau de prix de Lucentis (cf. paragraphes 858 à 861 de la présente décision). En effet, dans la mesure où il a eu pour effet de limiter les prescriptions d’Avastin (cf. paragraphes 839 à 856 ci-dessus), le discours de Novartis a pu avoir pour effet de reporter la date à laquelle le CEPS a pu considérer que, compte tenu de son utilisation courante par les médecins ophtalmologistes, Avastin devait être considéré comme un comparateur dans le traitement de la DMLA et pour les autres indications oculaires.
1165.Comme évoqué aux paragraphes 863 à 885 de la présente décision, le CEPS aurait pu procéder à une baisse du prix de Lucentis, d’abord, pendant la période de garantie de prix européen, qui s’étendait jusqu’en 2012, puis au moment de la renégociation du prix de Lucentis à l’issue de la période de garantie de prix européen.
1166.Il convient enfin de relever qu’en empêchant une réduction du prix de Lucentis, le discours véhiculé par Novartis a également conduit à fixer un niveau de prix d’Eylea artificiellement élevé : si le prix de Lucentis avait diminué avant l’entrée d’Eylea sur le marché, le prix de ce dernier aurait pu être fixé à un niveau inférieur, puisqu’il a été déterminé, selon les critères applicables aux spécialités dites « me-too », au même niveau que le prix de liste de Lucentis, minoré d’une décote de 10 % (cf. paragraphe 887 de la présente décision).
- Conclusion
1167.Il ressort des éléments analysés précédemment que les pratiques visées par le grief n° 1 ont causé un dommage significatif à l’économie.
Conclusion sur la proportion de la valeur des ventes
1168.Compte tenu de l’appréciation qu’elle a faite de la gravité des faits et de l’importance du dommage causé à l’économie dans le secteur concerné, l’Autorité retiendra, pour déterminer le montant de base de la sanction infligée à Novartis, une proportion de 14% comme assiette du montant de la sanction pécuniaire prononcée au titre du grief n° 1.
Sur la durée
Principes applicables
1169.Dans le cas d’infractions qui se sont prolongées plus d’une année, l’Autorité s’est engagée à prendre en compte leur durée selon les modalités pratiques suivantes : la proportion retenue, pour donner une traduction chiffrée à la gravité des faits et à l’importance du dommage causé à l’économie, est appliquée une fois, au titre de la première année complète de participation individuelle aux pratiques de chaque entreprise en cause, à la valeur de ses ventes pendant l’exercice comptable de référence, puis à la moitié de cette valeur, au titre de chacune des années complètes de participation suivantes. Au-delà de cette dernière année complète, la période restante est prise en compte au mois près, dans la mesure où les éléments du dossier le permettent.
1170.Dans chaque cas d’espèce, cette méthode se traduit par un coefficient multiplicateur, défini proportionnellement à la durée individuelle de participation de chacune des entreprises aux pratiques et appliqué à la proportion de la valeur des ventes effectuées par chacune d’entre elles pendant l’exercice comptable retenu comme référence.
Appréciation en l’espèce
1171.Au cas présent, comme cela ressort du paragraphe 907 de la présente décision, l’infraction sanctionnée au titre du grief n° 1 a débuté le 10 mars 2008, pour prendre fin au début du mois de novembre 2013.
1172.En conséquence, la durée des pratiques est de 5 ans et 7 mois, soit un coefficient multiplicateur de 3,29.
Conclusion sur la détermination du montant de base
1173.Eu égard à la gravité des faits et au dommage causé à l’économie par les pratiques sanctionnées au titre du grief n° 1, le montant de base de la sanction pécuniaire, déterminé en proportion des ventes en relation avec l’infraction réalisées par l’entreprise concernée, d’une part, et en fonction de la durée de l’infraction, d’autre part, est le suivant : 135 416 400 euros.
b) Sur l’individualisation de la sanction
1174.L’Autorité s’est ensuite engagée à adapter le montant de base retenu reflétant la gravité des faits et l’importance du dommage causé à l’économie au regard du critère légal tenant à la situation individuelle de l’entreprise sanctionnée et, quand c’est le cas, du groupe auquel elle appartient.
1175.À cette fin, et en fonction des éléments propres à chaque cas d’espèce, elle peut prendre en considération différentes circonstances atténuantes et/ou aggravantes caractérisant le comportement de l’entreprise mise en cause dans la commission des infractions, ainsi que d’autres éléments objectifs pertinents relatifs à sa situation individuelle. Cette prise en compte peut conduire à ajuster le montant de la sanction tant à la baisse qu’à la hausse.
En ce qui concerne les circonstances aggravantes et atténuantes
1176.Novartis soutient que la sanction prononcée à son encontre devrait être revue à la baisse compte tenu de l’existence de circonstances atténuantes. Plus spécifiquement, il fait valoir le contexte particulier dans lequel les pratiques se sont déroulées, ainsi que le caractère inédit des infractions sanctionnées, dont la qualification est complexe et ambigüe, justifierait qu’aucune sanction pécuniaire ne lui soit infligée ou, à tout le moins, le prononcé d’une amende symbolique.
1177.Toutefois, en premier lieu, les pratiques sanctionnées au titre du grief n° 1 ne revêtent, en tout état de cause, aucun caractère inédit. L’Autorité a déjà sanctionné, à plusieurs reprises, des pratiques abusives de dénigrement (cf. paragraphes 769 à 779 de la présente décision).
1178.En second lieu, les pratiques en cause se sont effectivement inscrites dans un contexte marqué par un débat scientifique sur l’efficacité et la sécurité d’utilisation d’Avastin « hors AMM » en ophtalmologie. Toutefois, un tel constat n’est pas de nature à justifier une réduction de la sanction prononcée, car il n’est pas reproché aux sociétés mises en cause d’avoir exprimé leur opinion dans ce contexte. Les pratiques sanctionnées reposent au contraire sur le fait que, loin de s’en tenir à un discours évoquant cette situation d’incertitude, la communication de Novartis avait pour objectif et a été de nature à renforcer les doutes sur l’usage « hors AMM » d’Avastin, en exagérant les risques qui y étaient associés, en comparaison avec l’usage autorisé de son propre produit, Lucentis.
1179.Il ressort de ce qui précède qu’aucune circonstance atténuante ne justifie de revoir le montant de la sanction prononcée au titre du grief n° 1 à la baisse. Par ailleurs, aucun élément du dossier ne permet de considérer que Novartis devrait voir le montant de sa sanction augmenté au titre de circonstances aggravantes.
En ce qui concerne les autres éléments d’individualisation
Principes applicables
1180.Les points 47 et 48 du communiqué sanctions rappellent à cet égard qu’« [a]fin d’assurer le caractère à la fois dissuasif et proportionné de la sanction pécuniaire, l’Autorité peut ensuite adapter, à la baisse ou à la hausse, le montant de base en considération d’autres éléments objectifs propres à la situation de l’entreprise ou de l’organisme concerné :
(…)
Elle peut aussi l’adapter à la hausse pour tenir compte du fait que :
- l’entreprise concernée dispose d’une taille, d’une puissance économique ou de ressources globales importantes, notamment par rapport aux autres auteurs de l’infraction ;
- le groupe auquel appartient l’entreprise concernée dispose lui-même d’une taille, d’une puissance économique ou de ressources globales importantes, cet élément étant pris en compte, en particulier, dans le cas où l’infraction est également imputable à la société qui la contrôle au sein du groupe ».
1181.En ce qui concerne l’adaptation à la hausse du montant de la sanction, il est de jurisprudence constante que l’appréciation de la situation individuelle peut conduire à prendre en considération l’envergure de l’entreprise en cause ou du groupe auquel elle appartient (voir en ce sens arrêt de la Cour de cassation du 28 avril 2004, Colas Midi-Méditerranée e.a. n° 02-15203).
1182.Ainsi, la Cour de justice, tout en indiquant que le recours à la valeur des ventes de l’entreprise en cause permet de proportionner l’assiette de la sanction à l’ampleur économique de l’infraction et au poids relatif de l’intéressée sur le secteur ou marché en cause, rappelle qu’il est légitime de tenir compte, dans le même temps, du chiffre d’affaires global de cette entreprise, en ce que celui-ci est de nature à donner une indication de sa taille, de sa puissance économique et de ses ressources (arrêts de la Cour de justice du 7 juin 1983 Musique Diffusion Française/Commission, C-100 à 103/80, points 119 à 121 ; du 26 juin 2006, Showa Denko/Commission, aff. C-289/04 P, Rec. p. I-5859, points 16 et 17 ; et du 4 septembre 2014, YKK Corporation, C-408/12, point 86).
1183.De fait, la circonstance qu’une entreprise dispose d’une puissance financière importante peut justifier que la sanction qui lui est infligée, en considération d’une ou plusieurs infractions données, soit plus élevée que si tel n’était pas le cas, afin d’assurer le caractère à la fois dissuasif et proportionné de la sanction pécuniaire (arrêt de la cour d’appel de Paris, 11 octobre 2012, Entreprise H. Chevalier Nord e.a. n° 2011-03298, p. 71, et du 30 janvier 2014, Société Colgate-Palmolive Service, n° 2012-00723, p. 41).
1184.À cet égard, la Cour de cassation a déjà eu l’occasion de préciser que l’efficacité de la lutte contre les pratiques anticoncurrentielles requiert que la sanction pécuniaire soit effectivement dissuasive – objectif également mis en exergue, s’agissant des sanctions pouvant être imposées en cas de violation de règles nationales de concurrence par la Cour européenne des droits de l’Homme (arrêt de la Cour européenne des droits de l’Homme du 27 septembre 2011, Menarini Diagnostics/Italie (req. n° 43509/08, point 41), au regard de la situation financière propre à chaque entreprise au moment où elle est sanctionnée (arrêt de la Cour de cassation du 18 septembre 2012, Séphora e.a, n° 12-14401).
1185.La cour d’appel de Paris l’a encore récemment rappelé dans un arrêt du 11 juillet 2019, société Janssen-Cilag SAS (arrêt de la cour d’appel de Paris du 11 juillet 2019, société Janssen-Cilag S.A.S e.a, n° 18/01945, points 581 et suivants). Elle a, en effet, précisé que la majoration du montant de base de la sanction en raison de l’appartenance à un groupe dépendait des circonstances de fait et du contexte propre à chaque espèce. Par ailleurs, elle a admis que cette puissance pouvait être révélée par le faible ratio entre la valeur des ventes retenues pour le calcul de l’assiette de la sanction et le chiffre d’affaires du groupe auquel appartient l’auteur de l’infraction.
Appréciation en l’espèce
1186.L’infraction sanctionnée au titre du grief n° 1 a été imputée aux sociétés Novartis Pharma SAS et Novartis AG, en tant qu’auteures, et aux sociétés Novartis Groupe France SA et Novartis AG, en tant que sociétés mères.
1187.Or, ces sociétés appartiennent au groupe Novartis, qui jouit d’une taille et d’une puissance économique importantes. Les ressources financières globales du groupe Novartis sont en effet très élevées. Son chiffre d’affaires consolidé hors taxes s’élève à 47 445 millions de dollars au 31 décembre 2019, soit 42 387 millions d’euros(52). Son résultat net consolidé s’établit à 11 737 millions de dollars la même année, soit 10 486 millions d’euros.
1188.La valeur des ventes retenue comme assiette de la sanction prononcée à son égard au titre du grief n° 1 représente ainsi seulement 0,69 % du chiffre d’affaires total du groupe et environ 2,8 % du résultat net consolidé.
1189.Compte tenu de ces éléments, le montant de base de la sanction pécuniaire infligée aux sociétés Novartis Pharma SAS et Novartis AG, solidairement avec leurs sociétés mères, Novartis Groupe France SA et Novartis AG, doit être augmenté de 50 %.
1190.Eu égard à l’ensemble des éléments qui précèdent, le montant de la sanction pécuniaire à imposer aux sociétés Novartis Pharma SAS et Novartis AG, conjointement et solidairement avec les sociétés Novartis Groupe France SA et Novartis AG, sera fixé à 203 124 600 euros au titre du grief n° 1.
c) Sur la réitération
Principes applicables
1191.La réitération, visée de façon autonome par le I de l’article L. 464-2 du code de commerce, constitue une circonstance aggravante que l’Autorité peut prendre en compte en augmentant le montant de la sanction de manière à lui permettre d’apporter une réponse proportionnée, en termes de répression et de dissuasion, à la propension de l’entreprise ou de l’organisme de s’affranchir des règles de concurrence (décision n° 07-D-33 du Conseil du 15 octobre 2007 relative à des pratiques mises en œuvre par la société France Télécom dans le secteur de l’accès à Internet à haut débit, paragraphe 112). La jurisprudence de l’Union va dans le même sens (arrêt de la Cour de justice du 8 février 2007, Groupe Danone/Commission, C-3/06 P, Rec. p. I-1331, point 47).
1192.Il est possible de retenir l’existence d’une réitération lorsque quatre conditions sont réunies (paragraphe 51 du communiqué du 16 mai 2011) : une précédente infraction au droit de la concurrence doit avoir été constatée avant la fin de la commission de la nouvelle pratique ; la nouvelle pratique doit être identique ou similaire, par son objet ou ses effets, à celle ayant donné lieu au précédent constat d’infraction ; ce dernier doit avoir acquis un caractère définitif à la date à laquelle l’Autorité statue sur la nouvelle pratique ; le délai écoulé entre le précédent constat d’infraction et le début de la nouvelle pratique est pris en compte pour apporter une réponse proportionnée à la propension de l’entreprise à s’affranchir des règles de concurrence, étant indiqué que la réitération ne sera ainsi pas retenue lorsque le délai en question est supérieur à quinze ans.
1193.Selon la jurisprudence, la qualification de la réitération n'exige pas que les infractions commises soient identiques quant à la pratique mise en œuvre ou quant au marché concerné, qu'il s'agisse du marché de produits ou services ou du marché géographique, et qu'elle peut être retenue pour de nouvelles pratiques identiques ou similaires, par leur objet ou leurs effets, à celles ayant donné lieu au précédent constat d'infraction (arrêt de la cour d’appel de Paris du 4 juillet 2013, n° 2012/05160, page 35, confirmé par un arrêt de la Cour de cassation du 6 janvier 2015, n° 13-21305 et 13-22477).
1194.Enfin, les règles en matière de réitération doivent suivre celles appliquées en matière d'imputabilité. En conséquence, il doit être tenu compte, pour apprécier cette circonstance aggravante, du fait que l'une des personnes morales composant l'entreprise en cause, au sens des articles 101 et 102 TFUE, a déjà été sanctionnée pour avoir commis une infraction de même type (arrêt de la cour d’appel de Paris du 4 juillet 2013, n° 2012/05160, page 35, confirmé par un arrêt de la Cour de cassation du 6 janvier 2015, Orange Caraïbe, n° 13-21305 et 13-22477).
1195.Plus spécifiquement, la Cour de justice a jugé que « pour établir la circonstance aggravante de récidive à l’égard de la société mère, il n’est pas exigé que cette dernière ait fait l’objet de poursuites antérieures, ayant donné lieu à une communication des griefs (…) ce qui importe notamment est la constatation antérieure d’une première infraction résultant du comportement d’une filiale avec laquelle la société mère impliquée dans la second infraction formait, déjà au moment de la première infraction, une seule entreprise » (arrêt du 5 mars 2015, Versalis SpA et Enin SpA, C-93/13, point 31).
Appréciation en l’espèce
1196.La société Novartis Pharma SAS a été sanctionnée par une décision du Conseil de la concurrence pour des pratiques de remises fidélisantes à l’hôpital, constitutives d’abus de position dominante, devenue définitive le 28 juin 2005 (décision n° 03-D-35 du 24 juillet 2003 relative à des pratiques mises en œuvre par les laboratoires Sandoz, devenus en 1997 Novartis Pharma SA, sur le marché de certaines spécialités pharmaceutiques destinées aux hôpitaux, confirmée par un arrêt de la cour d’appel de Paris du 30 mars 2004 et un arrêt de la Cour de cassation du 28 juin 2005, Novartis Pharma, n° 04-13910).
1197.Dans la présente décision, il est établi que Novartis Pharma SAS a mis en œuvre, en tant qu’auteur, postérieurement à ce constat d’infraction, des pratiques constitutives d’abus de position dominante collective à compter du 10 mars 2008 s’agissant du grief n° 1, d’une part, et à compter du 9 mai 2011 s’agissant du grief n° 2, d’autre part. Novartis Pharma SAS a donc mis en œuvre des pratiques similaires à celles sanctionnées dans la décision n° 03-D-35, qui sont intervenues cinq ans (s’agissant du grief n° 1) et huit ans (s’agissant du grief n° 2) après le constat d’infraction du comportement de Novartis Pharma SA aux règles de concurrence par la décision datée de juillet 2003.
1198.En conséquence, il y a lieu de constater que la société Novartis Pharma SAS se trouve dans une situation de réitération, ce qui du reste, n’est pas contesté par Novartis. Il convient donc de retenir, dans les circonstances de l’espèce, une majoration de 25 % de la sanction prononcée à l’encontre de Novartis Pharma, portant celle-ci à un montant de 253 905 750 euros au titre du grief n° 1.
1199.S’agissant de l’implication de Novartis AG en tant qu’auteur des pratiques, il n’est pas établi qu’elle existait à la date du premier constat d’infraction. Dès lors, il y a lieu de constater que cette société ne se trouve pas en situation de réitération.
1200.De même, s’agissant de la responsabilité de Novartis AG et de Novartis France Groupe SA en tant que sociétés mères, il n’est pas établi qu’elles formaient, à la date du premier constat d’infraction, avec la société Novartis Pharma SAS, une seule entreprise au sens des articles 101 et 102 du TFUE. Dès lors, il y a lieu de constater que ces sociétés ne se trouvent pas en situation de réitération.
1201.Il résulte de ce qui précède que (i) la sanction prononcée à l’encontre de Novartis au titre du grief n° 1 sera de 253 905 750 euros et (ii) Novartis Pharma SAS sera tenue seule responsable de la majoration de la sanction prononcée au titre de la réitération, correspondant au montant suivant : 50 781 150 euros.
d) Conclusion sur le montant de la sanction pécuniaire
1202.Au vu des considérations qui précèdent, il y a lieu d’imposer à Novartis, au titre du grief n° 1, une sanction pécuniaire d’un montant de 253 905 750 euros, étant précisé que Novartis Pharma SAS sera tenue seule responsable de cette sanction à hauteur de 50 781 150 euros.
e) Sur le maximum légal
1203.Lorsque les comptes sont consolidés, le plafond légal de la sanction correspond à 10 % du chiffre d’affaires mondial hors taxes consolidé le plus élevé réalisé au cours d’un des exercices clos depuis l’exercice précédant le commencement des pratiques.
1204.Le chiffre d’affaires mondial consolidé hors taxes le plus élevé connu réalisé par le groupe Novartis était de 47 445 millions de dollars au 31 décembre 2019 euros, soit environ 42 387 millions d’euros(53).
1205.Le montant de sanction retenu précédemment étant inférieur à 10 % de ce chiffre, il n’y a pas lieu de le modifier.
2. LE GRIEF N° 2
a) Sur la détermination du montant de base des sanctions pécuniaires
Sur la valeur des ventes
1206.L’Autorité a rappelé aux paragraphes 1119 à 1122 de la présente décision les modalités de détermination de la valeur des ventes.
1207.En l’espèce, l’infraction sanctionnée au titre du grief n° 2 – dont la durée de participation diffère selon les sociétés concernées – concerne les ventes de médicaments pour le traitement de la DMLA et d’autres pathologies oculaires (OMD, OBVR, OVCR ou baisse visuelle due à une NVC) sur le territoire français.
Sur l’assiette de la sanction pécuniaire de Roche et Genentech
1208.Comme souligné au paragraphe 1213 ci-dessous, le groupe Roche réalise des ventes en lien avec l’infraction au travers de sa seule filiale Genentech.
1209.Pour les raisons évoquées au paragraphe 1125 de la présente décision, le prononcé d’une sanction pour chaque société du groupe Roche est inadapté.
1210.En conséquence, la sanction pécuniaire prononcée à l’encontre du groupe Roche sera assise sur une seule valeur des ventes, correspondant au chiffre d’affaires de la société du groupe commercialisant des produits en relation avec l’infraction, c’est-à-dire celui de Genentech (cf. paragraphes 1213 à 1218 ci-dessous).
Sur le périmètre de la valeur des ventes
1211.Il convient de retenir les ventes en lien avec l’infraction, c'est-à-dire les ventes de médicaments pour le traitement de la DMLA et d’autres pathologies oculaires (OMD, OBVR, OVCR ou baisse visuelle due à une NVC) sur le territoire français, tant en ville qu’à l’hôpital.
1212.S’agissant de Novartis, ces ventes correspondent aux ventes de Lucentis en France.
1213.S’agissant des entités du groupe Roche, celui-ci a communiqué aux services d’instruction des estimations des ventes totales d’Avastin en France. Mais eu égard à la nature des pratiques litigieuses, qui visaient à limiter l’utilisation d’Avastin en ophtalmologie sur le territoire français, la prise en compte de ces ventes aboutirait à un résultat ne reflétant manifestement pas de façon appropriée l’ampleur économique de l’infraction. Conformément au point 39 du communiqué sanctions précité, il appartient alors à l’Autorité de rechercher une assiette alternative pour le calcul du montant de base de la sanction pour les entités du groupe Roche.
1214.Il apparaît que le groupe Roche perçoit deux principales sources de revenus en lien avec les pratiques. D’une part, Genentech perçoit des redevances sur les ventes de Lucentis. D’autre part, Genentech vend à Novartis le principe actif de Lucentis pour sa commercialisation sur le territoire français.
1215.Roche soutient que les revenus de Genentech ne peuvent être retenus, sauf à considérer que cette entité occupe un rôle prépondérant dans la commission des pratiques reprochées, ce qui ne serait pas le cas en l’espèce. Mais il ressort des paragraphes 1018 à 1028 de la présente décision que Genentech a participé à l’infraction en qualité d’auteur, ce qui justifie de prendre en considération son chiffre d’affaires.
1216.Les redevances perçues par Genentech ont été communiquées aux services d’instruction. Il s’agit des redevances perçues auprès de Novartis d’une part, et de Roche SAS d’autre part. Pour ces dernières, Roche propose deux estimations différentes, la première fondée sur les données collectées via le programme de médicalisation des systèmes d’information, la deuxième sur les données issues des rapports de suivi de mise en œuvre de la RTU d’Avastin. Ces deux estimations faisant apparaître des montants comparables, il sera retenu la plus faible des deux. En revanche, Roche a indiqué ne pas être en mesure de distinguer les ventes du principe actif de Lucentis selon le pays de destination du produit fini. Il appartient dans ces conditions à l’Autorité, et conformément au point 36 du communiqué sanctions susmentionné, de se déterminer à partir des données dont elle dispose.
1217.En l’espèce, l’Autorité relève, d’une part, que Roche a communiqué les valeurs des ventes du principe actif de Lucentis sur un périmètre monde, à l’exclusion des États-Unis (cote 50357). D’autre part, Novartis a communiqué aux services d’instruction la répartition des volumes de ventes de Lucentis en France et dans le reste du monde (hors États-Unis) (cote 50362). À défaut d’informations plus précises, il sera considéré que la part des volumes de ventes de Lucentis réalisée en France (soit un pourcentage de 16,6 %), appliquée aux ventes totales du principe actif de Lucentis, reflète de manière appropriée le produit des ventes du principe actif de Lucentis en France par Genentech.
1218.En conséquence, il convient de retenir comme valeur des ventes en relation avec l’infraction, les redevances perçues par Genentech sur les ventes de Lucentis en France, d’une part, et les ventes par Genentech de principe actif de Lucentis pour sa commercialisation sur le territoire français, d’autre part.
Sur l’exercice de référence
1219.Le dernier exercice complet de participation de Novartis, Roche et Genentech à l’infraction est l’exercice 2012.
Conclusion
1220.Il ressort de ce qui précède que l’assiette sur laquelle seront assises les sanctions pécuniaires prononcées au titre du grief n° 2 est la suivante :
| Sociétés | Valeur des ventes (en euros) |
| Novartis Pharma SAS et Novartis AG | […] |
| Roche/Genentech(54) | […](55) |
Sur la proportion de la valeur des ventes
Sur la gravité des pratiques
1221.L’Autorité a rappelé aux paragraphes 1132 et 1133 de la présente décision les modalités de prise en compte de la gravité des pratiques.
1222.En l’espèce, il ressort des développements qui précèdent que Roche et Novartis ont, avec l’appui de Genentech, développé et diffusé un discours alarmiste, parfois trompeur, auprès des autorités publiques sur les risques liés à l’utilisation d’Avastin pour le traitement de la DMLA, afin de bloquer ou ralentir, de façon indue, les initiatives des pouvoirs publics qui envisageaient de favoriser et sécuriser son usage pour le traitement de la DMLA.
1223.Les pratiques en cause sont particulièrement graves, car elles sont intervenues dans le secteur de la santé, et plus spécifiquement, dans un contexte de débat public sur le prix extrêmement élevé de Lucentis et sur son impact sur les finances sociales, pour lesquelles le remboursement à 100 % de Lucentis constituait un poste de dépense significatif, alors qu’il existait un médicament, Avastin, nettement moins cher, susceptible d’être utilisé en ophtalmologie.
1224.Or, les pratiques en cause visaient non seulement à empêcher que l’utilisation d’Avastin « hors AMM » pour le traitement de la DMLA, et plus généralement en ophtalmologie, par les médecins ophtalmologistes soit facilitée par une prise de position des autorités publiques, mais également à éviter qu’Avastin ne soit considéré comme comparateur de Lucentis. En effet, plusieurs documents internes de Novartis démontrent l’inquiétude du laboratoire concernant l’impact que la concurrence d’Avastin pouvait avoir sur le prix de Lucentis (cf. paragraphes 259 à 260, 291 et 359 de la présente décision).
1225.Par ailleurs, le nombre, le cumul et l’interaction des comportements anticoncurrentiels mis en œuvre en même temps constituent un facteur qui doit être pris en compte au titre de la gravité des faits.
1226.Or, en l’espèce, le discours adressé par Roche, d’une part, et par Novartis, d’autre part, aux autorités publiques, a été complémentaire et s’est cumulé, dans un but commun de nuire à l’utilisation d’Avastin « hors AMM » pour le traitement de la DMLA, et plus généralement en ophtalmologie, et de maintenir le prix de Lucentis à un prix artificiellement élevé. Ainsi, non seulement le laboratoire Roche ne s’est pas opposé au discours véhiculé par Novartis concernant le médicament qu’il commercialise, mais il est établi qu’il a adopté une communication similaire à celle de son concurrent, en faveur de l’usage de Lucentis et, de fait, au détriment des ventes de son propre produit. En outre, l’implication de Genentech a permis à Roche et Novartis de développer et diffuser un discours similaire.
1227.Les mises en cause soutiennent toutefois que la gravité des pratiques est d’une ampleur limitée. Plus spécifiquement, Roche estime qu’il devrait être tenu compte du fait que les pratiques en cause présenteraient un caractère inédit, que ses démarches vis-à-vis des autorités publiques ont été passives et limitées, ou encore que Genentech n’est jamais intervenu directement auprès des autorités publiques. Par ailleurs, Roche et Novartis soulignent que les pratiques n’auraient été structurées que sur une très courte période. Enfin, Roche et Novartis prétendent, au titre de l’appréciation de la gravité, que leur discours n’était pas trompeur, mais justifié par des considérations de santé publique.
1228.Toutefois, en premier lieu, les pratiques sanctionnées au titre du grief n° 2 ne revêtent aucun caractère inédit. En effet, à la date des faits, avaient déjà été sanctionnées comme constitutives d’abus de position dominante, des pratiques d’intervention auprès d’une autorité publique, que les informations communiquées à l’autorité publique soient ou non trompeuses (voir la décision n° 05-D-58 du 3 novembre 2005 relative à des pratiques relevées dans le secteur de l’eau potable en Ile-de-France et la décision de la Commission européenne du 15 juin 2005, AstraZeneca, COMP/A. 37.507). La cour d’appel a jugé en ce sens que « Rien, dans ces décisions n’indique que l’intervention auprès d’une autorité publique n’est susceptible de constituer une pratique anticoncurrentielle que si l’opérateur économique communique à cette autorité des informations trompeuses » et que « depuis ces décisions, toute entreprise en position dominante est avertie que son intervention dans le processus décisionnel d’une autorité publique peut, en fonction des circonstances de l’espèce, être qualifiée d’abus de position dominante » (arrêt de la cour d’appel de Paris du 11 juillet 2019, société Janssen-Cilag SAS, n° 18/01945, points 446 et 447).
1229.En deuxième lieu, s’il est exact que les comportements poursuivis ont consisté, pour l’essentiel, en un ensemble de contacts ponctuels de Roche et Novartis avec les autorités publiques, leur nombre, leur cumul et leur interaction, tels qu’exposés ci-dessus, leur confère, au contraire, un degré de sophistication certain.
1230.En troisième lieu, le prétendu caractère limité de l’intervention de Roche et de Genentech ne saurait être apprécié au titre de la gravité des pratiques, une telle circonstance relevant de l’individualisation des sanctions.
1231.En dernier lieu, il est établi que les pratiques en cause n’étaient justifiées par aucun objectif de protection de la santé publique. En effet, l’examen du discours de Roche et de Novartis, établi avec le soutien de Genentech, révèle la ligne d’action commune dégagée par l’entité collective, qui n’est pas justifiée par des considérations de santé publique, et constitue une pratique étrangère à la concurrence par les mérites.
1232.Dès lors, les arguments des mises en cause seront écartés.
1233.Il ressort de ce qui précède que les pratiques sanctionnées au titre du grief n° 2 doivent être considérées comme très graves.
Sur le dommage causé à l’économie
1234.L’Autorité a rappelé aux paragraphes 1143 à 1145 de la présente décision les modalités de prise en compte du dommage causé à l’économie.
- Sur l’ampleur des pratiques
1235.L’ampleur du marché affecté est particulièrement importante dès lors que les pratiques en cause, consistant à faire obstacle aux initiatives des pouvoirs publics français pour encourager l’utilisation d’Avastin « hors AMM », ont été déployées par les sociétés mises en cause, auprès de plusieurs institutions publiques, en particulier les autorités du secteur de la santé (telles que l’AFSSAPS/l’ANSM, la HAS, le CEPS, la DGS ou le cabinet du ministre de la santé), mais également d’autres acteurs institutionnels (comme la présidence de la République, les services du Premier ministre ou encore le ministère de l’économie).
1236.De plus, Genentech, Roche et Novartis détenaient une position dominante collective sur le marché français de la DMLA exsudative, couvrant la totalité des anti-VEGF utilisés en injection intravitréennes, jusqu’à l’arrivée d’Eylea. De ce fait, aucun concurrent sur le marché ne pouvait ou n’était incité à s’opposer aux pratiques mises en œuvre par les laboratoires membres de l’entité collective.
1237.Par ailleurs, toutes les indications ophtalmologiques d’Avastin ont été affectées par la pratique (cf. paragraphe 1055 de la présente décision).
1238.Roche considère, dans son mémoire en réponse au rapport, que l’ampleur alléguée des pratiques est principalement imputable à Novartis, et fait valoir que le poids de Novartis sur le marché est considérablement plus important.
1239.Néanmoins, comme souligné au paragraphe 1034 de la présente décision, Roche ne s’est pas opposé au discours véhiculé par Novartis auprès des autorités publiques, mais, à l’inverse, a instrumentalisé les risques d’effets secondaires liés à l’utilisation d’Avastin « hors AMM » en ophtalmologie pour, dans un premier temps, « justifier » son refus de communiquer les éléments demandés par l’AFSSAPS en 2008 et 2009, retardant ainsi la réalisation de l’étude de comparaison GEFAL (cf. paragraphes 925 à 938 de la présente décision), et dans un second temps, retarder les initiatives des pouvoirs publics visant à encadrer et sécuriser l’utilisation d’Avastin en ophtalmologie (cf. paragraphes 942 à 974 de la présente décision).
1240.Ainsi, d’abord, les effets du comportement de Roche et son rôle dans l’ampleur de la pratique sont indépendants du poids relatif du laboratoire sur le marché concerné par rapport à Novartis. Ensuite, dans la mesure où les pratiques en cause visaient à limiter l’usage d’Avastin en ophtalmologie, la prise en compte du poids de Roche sur le marché concerné n’est pas pertinente. Enfin, et en tout état de cause, d’une part, le dommage causé par une pratique anticoncurrentielle s’apprécie de manière globale, pour l’ensemble des participants et, d’autre part, le poids respectif de Novartis et de Roche est pris en compte dans le cadre de la détermination de la valeur des ventes.
- Sur les caractéristiques du secteur
1241.Les caractéristiques économiques du secteur en cause amplifient l’importance du dommage à l’économie.
1242.Tout d’abord, le dommage à l’économie est particulièrement important en raison du différentiel de coût d’Avastin (environ 30 euros par injection) et de Lucentis (plus de 1 000 euros par injection) (cf. paragraphe 1155 de la présente décision).
1243.Par ailleurs, le marché est caractérisé par de forts coûts de développement constitutifs de barrières à l’entrée élevées, rendant ainsi plus difficile l’entrée de nouveaux concurrents (cf. paragraphe 1156 de la présente décision).
1244.De plus, l’aversion au risque des professionnels de santé, en particulier des représentants des autorités de santé, en raison notamment d’une certaine judiciarisation des questions de santé, a pour conséquence que toute contestation, fondée ou non, de la part de Novartis et de Roche de la sécurité d’utilisation de leurs médicament conduit quasi-inéluctablement à un ralentissement du processus décisionnel des autorités publiques. Partant, toute exagération des risques liés à l’utilisation d’Avastin en ophtalmologie ne pouvait qu’avoir un effet sensible sur les autorités de santé, soucieuses de protéger les intérêts des patients, et les inciter à recommander aux professionnels de santé de privilégier Lucentis au détriment d’Avastin.
1245.Enfin, compte tenu des modalités de négociation du prix du médicament entre le CEPS et le laboratoire titulaire, présentées aux paragraphes 30 à 43 de la présente décision, une pratique visant, comme celle en cause au titre du grief n° 2, à retarder les initiatives des pouvoirs publics pour encadrer et sécuriser l’utilisation « hors AMM » d’un médicament comme Avastin, est susceptible d’avoir un impact important sur le coût pour les finances sociales de cette catégorie de traitement. En effet, la reconnaissance par les autorités publiques de l’équivalence thérapeutique et de la sécurité d’usage d’un médicament comme Avastin constituait un élément susceptible d’être pris en compte dans le cadre des négociations du prix réglementé de Lucentis.
- Sur les conséquences structurelles et conjoncturelles
1246.Les comportements reprochés à Novartis, Roche et Genentech ont eu un impact sur les autorités publiques et, partant, sur l’utilisation « hors AMM » d’Avastin en ophtalmologie. Ils recouvrent différents aspects, qui ont tous eu des conséquences conjoncturelles et structurelles.
1247.Tout d’abord, le refus de Roche de communiquer les données scientifiques à l’AFSSAPS en 2008, puis en 2009, a directement contribué au retard, d’au moins 16 mois, dans la mise en place de l’étude GEFAL, dont les résultats n’ont été rendus publics qu’en mai 2013. Ce refus a donc, in fine, contribué au ralentissement de la régularisation de l’utilisation d’Avastin en ophtalmologie.
1248.Ensuite, entre 2011 et 2013, Roche et Novartis ont diffusé auprès des pouvoirs publics (ministère de la santé) et des autorités de santé (AFSSAPS/ANSM, CEPS, HAS) un discours alarmiste, voire trompeur, sur les risques d’effets secondaires graves pour les patients traités avec Avastin en injection intravitréenne, notamment en interprétant de manière orientée les résultats publiés des études de comparaison. Genentech a permis de coordonner le discours de Novartis et de Roche concernant les deux spécialités et, partant, de définir une ligne d’action commune entre les trois laboratoires, de façon à ce que Roche et Novartis puissent développer un discours très similaire, fondé sur les mêmes éléments techniques.
1249.Les conséquences de ce discours sont attestées de trois manières.
1250.Premièrement, comme évoqué précédemment, même s’il peut exister certains freins, indépendants des pratiques, à l’utilisation « hors AMM » d’Avastin (i.e., nécessité d’un équipement adéquat pour le reconditionnement stérile d’Avastin, obligation d’information des patients par les prescripteurs, péremption des seringues d’Avastin, recours d’Avastin devant être considéré comme indispensable par le médecin prescripteur), les pièces du dossier confirment qu’en 2011, en dépit du comportement de Novartis visé par le grief n° 1, Avastin était bien utilisé en dehors de son AMM par une partie des praticiens français (paragraphe 180 de la décision). Les pouvoirs publics envisageaient à cet égard, malgré le choix fait par Roche de ne pas développer Avastin pour une indication en ophtalmologie, et compte tenu de cette pratique répandue des ophtalmologues français, de favoriser et sécuriser son usage « hors AMM » en ophtalmologie.
1251.Deuxièmement, comme évoqué au paragraphe 1037 de la présente décision, certains documents internes de Novartis, comme par exemple le compte rendu rédigé par le directeur de la « Franchise Ophtalmologie » de Novartis, en vue de son évaluation individuelle pour l’année 2013, montrent que le discours de dénigrement véhiculé par les mises en cause auprès des autorités de santé a eu un réel effet sur les opinions des responsables publics de la santé en France : « First official Novartis written communication clearly stating Lucentis vs Avastin safety différence, in coopération with top KOLs. Awareness of these safety issues has really increased in France (cf. Gefal communications, Pr Marananchi audition to senate, CNAM report to Assemblée Nationale, general public media articles,...) which can explain that RTU décret is still not published and that Health Authorities bodies are reorienting their actions towards a Lucentis price discussion more than an Avastin endorsement », (traduction libre : « Première communication écrite officielle de Novartis indiquant les différences entre Lucentis et Avastin en termes de sécurité, en coopération avec les meilleurs KOLs [leaders d’opinion]. L’attention sur ces questions de sécurité a réellement progressé en France (cf. communication Gefal, articles dans les médias…), ce qui peut expliquer que le décret RTU n’est toujours pas publié et que les autorités de santé réorientent leurs actions vers une discussion du prix de Lucentis, plutôt qu’une approbation d’Avastin », cote 45126).
1252.Troisièmement, les prises de position officielles des autorités publiques montrent également que le discours véhiculé par les laboratoires pharmaceutiques mis en cause a eu un effet sur les opinions des responsables publics français. À titre d’illustration, dans son point d’information du 16 septembre 2011, l’AFSSAPS a relayé les réserves de Roche sur la méthodologie de l’étude CATT et sur la tolérance d’Avastin lors d’une utilisation « hors AMM » en ophtalmologie (cote 49619).
1253.De même, les pratiques en cause ont directement contribué à l’adoption de l’instruction de la DGS de juillet 2012 (cf. paragraphe 973 de la présente décision).
1254.Ensuite, le discours de Roche et de Novartis, avec le soutien de Genentech, visait également à retarder l’adoption des dispositions législatives établissant le principe d’une RTU pour l’usage d’Avastin pour le traitement de la DMLA (cf. paragraphes 1044 à 1050 de la présente décision).
1255.Or, à partir du moment où les pouvoirs publics ont annoncé étudier la possibilité d’une RTU pour Avastin pour le traitement de la DMLA, Roche et Novartis ont multiplié les initiatives et les contacts avec les représentants du Gouvernement et les autorités de santé pour diffuser leurs messages concernant les risques associés à l’utilisation de cette spécialité en intravitréen (cf. paragraphes 470 et suivants de la présente décision). Ces comportements se sont ensuite poursuivis tout au long des débats parlementaires sur la réforme du mécanisme des RTU proposée dans le PLFSS 2013 (cf. paragraphes 535 à 544 de la présente décision).
1256.Les pièces au dossier montrent ainsi que l’autorité française de santé a cherché, dès février 2007, à prendre position sur l’usage d’Avastin « hors AMM » par les médecins ophtalmologistes, jusqu’alors non encadré. En effet, en février 2007, l’AFSSAPS annonçait engager une réflexion sur la mise en place d’un PTT pour cette spécialité, mais souhaitait disposer d’une étude comparative entre Lucentis et Avastin (cf. paragraphes 432 à 435 de la présente décision). Quatre ans plus tard, au mois de mai 2011, soit au moment de la publication des résultats des études Gower et al et CATT, l’AFSSAPS proposait à Roche la mise en place d’un PTT pour sa spécialité, compte tenu de l’enrichissement du corpus scientifique et de la pratique clinique des ophtalmologistes (cf. paragraphes 451 et 452 de la présente décision). Les pratiques de Roche, Novartis et Genentech s’inscrivent donc, en 2008 (s’agissant des pratiques reprochées à Roche et Genentech) et en 2011 (s’agissant des pratiques reprochées à Roche, Genentech et Novartis), dans le débat public en cours sur l’utilisation « hors AMM » d’Avastin en ophtalmologie.
1257.Par conséquent, le discours de Roche et Novartis, avec l’aide de Genentech a directement contribué au retard de l’adoption des dispositions législatives nécessaires à l’encadrement et la sécurisation de l’usage d’Avastin en ophtalmologie.
1258.L’ensemble de ces pratiques ont alors eu deux types d’effets sur le marché.
1259.En premier lieu, ces pratiques ont conduit à diminuer durablement la part de marché d’Avastin sur le marché du traitement de la DMLA. Il peut ainsi être considéré que, sur la base des données de sondage de Novartis et compte tenu des pratiques de Novartis déjà sanctionnées au titre du grief n° 1, les parts de marché d’Avastin sur la période 2007-2012 se situent en moyenne autour de 5 % (cote 4705). Par la suite, l’instruction de la DGS de juillet 2012, à laquelle les pratiques ont contribué, rend l’utilisation d’Avastin pour les traitements ophtalmologiques quasiment nulle (cf. paragraphe 180 de la présente décision). Par ailleurs, les pratiques sanctionnées au titre du grief n° 2 ont retardé le retour de la part de marché d’Avastin à son niveau antérieur aux pratiques visées tant par le premier grief que par le second grief, soit, à titre conservateur, une part de marché qui, compte tenu du discours déployé par Novartis et visé au premier grief, est de l’ordre de 5 % en moyenne sur la période 2007-2012. Or, l’adoption de dispositions législatives et réglementaires permettant d’encadrer et de sécuriser l’utilisation d’Avastin en ophtalmologie aurait pu accroître les parts de marché d’Avastin au-dessus de ce niveau.
1260.À l’inverse, les mises en cause considèrent quant à elles que la période suivant l’obtention de la RTU de 2015 est la meilleure période de référence pour évaluer l’impact éventuel des pratiques. À cet égard, l’analyse des autres médicaments ayant fait l’objet d’une RTU entre 2014 et 2016 montrerait que ce type d’autorisation n’a eu aucun impact sur l’utilisation de ceux-ci. Néanmoins, les arguments des mises en cause ne sont pas probants pour les raisons déjà évoquées aux paragraphes 851 à 855 de la présente décision.
1261.Par ailleurs, contrairement aux assertions de Novartis et Roche, selon lesquels il était impossible d’adopter une RTU pour Avastin avant décembre 2014 et les pratiques n’ont suscité aucun effet d’inertie après novembre 2013, la durée de ces effets a pu être particulièrement longue, allant, approximativement de 2011 à septembre 2015.
1262.En effet, comme indiqué supra, les pratiques ont contribué, d’une part, à l’adoption de l’instruction de la DGS de juillet 2012, et d’autre part, au retard de l’adoption des dispositions législatives nécessaires à l’établissement d’une RTU pour Avastin dans le traitement de la DMLA et, plus généralement au retard de l’adoption de dispositions permettant d’encadrer et de sécuriser l’utilisation d’Avastin « hors AMM » en ophtalmologie. Or, s’il est difficile de déterminer avec précision la date à laquelle ces dispositions auraient pu être adoptées en l’absence des pratiques, il peut être relevé que les pratiques reprochées et participant à ce retard débutent dès 2008, avec le refus de Roche de communiquer des données scientifiques à l’AFSSAPS. S’agissant du terme de ces effets, les pratiques ont participé au retard dans l’adoption d’une RTU pour l’utilisation « hors AMM » d’Avastin dans le traitement de la DMLA au moins jusqu’à l’adoption de la RTU dans la LFSS 2013 votée en décembre 2012. Si le retard ultérieur dans l’adoption de cette RTU n’est pas imputable aux mises en cause, les pratiques visées ont, en revanche, contribué à l’adoption de l’instruction de la DGS de juillet 2012, qui ne sera abrogée qu’en septembre 2015 avec la publication de la RTU d’Avastin. Ainsi, du fait des pratiques visées par ce grief, les praticiens n’ont pu prescrire Avastin dans le traitement de la DMLA que très difficilement et de façon marginale jusqu’à la publication de la RTU, en septembre 2015.
1263.En second lieu, par leurs pratiques, Roche, Novartis et Genentech ont également rendu plus difficile, pour les autorités de santé, la possibilité de considérer Avastin comme un comparateur pertinent de Lucentis, ce qui, à nouveau, auraît pu entrainer des demandes de baisses significatives du prix de Lucentis, et par voie de conséquence, un prix d’Eylea inférieur à son niveau réel (cf. paragraphe 1065 de la présente décision).
1264.Ainsi, comme vu précédemment, en l’absence des pratiques visées par le grief n° 1, le prix de Lucentis aurait pu baisser au cours de la période de garantie de prix européen, du fait de l’utilisation « hors AMM » d’Avastin par les praticiens, et ce même si les autorités de santé n’étaient pas encore certaines de l’équivalence thérapeutique à cette époque (cf. paragraphes 149 à 164 de la présente décision) et, a fortiori, ensuite, au fur et à mesure que les études scientifiques (Curtis et al, en octobre 2010, Gower et al. en mai 2011 ; CATT, en mai 2011 ; IVAN, en avril 2012 et GEFAL, en mai 2013, après un retard d’environ 16 mois du fait du refus de Roche de communiquer les données scientifiques), confirmaient l’équivalence thérapeutique et la tolérance d’Avastin en ophtalmologie, pouvant justifier que ce médicament soit pris comme comparateur.
1265.À cet égard, comme évoqué aux paragraphes 668 à 673 de la décision, Novartis s’inquiétait des résultats des études de comparaison et anticipait la prise en compte d’Avastin comme comparateur par les autorités publiques, au premier plan desquelles le CEPS.
1266.Or, dans les mois qui ont suivi la publication de l’avis de la HAS, en octobre 2017, établissant formellement qu’Avastin, dans le cadre d’une RTU, est un comparateur cliniquement pertinent de Lucentis, le CEPS a négocié et obtenu une baisse de 15 % du prix facial de Lucentis en octobre 2018. Comme l’indiquent les parties, la diminution de prix obtenue en 2018 peut, au moins en partie, résulter d’autres facteurs (extension des indications de Lucentis, changement du gouvernement et effort demandé à plusieurs industriels de la santé). De plus, comme indiqué supra, les pratiques reprochées au titre du grief n° 1, en diminuant artificiellement la part de marché d’Avastin, ont également pu empêcher le CEPS de procéder à des baisses de prix.
1267.Néanmoins, la concomitance entre l’avis précité de la HAS et la baisse de prix obtenue par le CEPS démontre qu’une baisse de prix de plusieurs points de pourcentage aurait pu intervenir une fois les résultats des études publiés et pris en compte par les autorités de santé françaises, soit, approximativement, en 2011.
1268.Enfin, une telle baisse de prix, qui aurait pu être imposée avant l’entrée d’Eyléa sur le marché en 2013, aurait également entraîné un prix d’Eylea inférieur.
- Conclusion
1269.Il ressort des éléments analysés précédemment que les pratiques poursuivies par le grief n° 2 ont causé un dommage significatif à l’économie.
Conclusion sur la proportion de la valeur des ventes
1270.Compte tenu de l’appréciation qu’elle a faite de la gravité des faits et de l’importance du dommage causé à l’économie dans le secteur concerné, l’Autorité retiendra, pour déterminer le montant de base des sanctions pécuniaires infligées aux entreprises en cause, une proportion de 14 % comme assiette du montant des sanctions pécuniaires prononcées au titre du grief n° 2.
Sur la durée
1271.L’Autorité a rappelé aux paragraphes 1169 et 1170 de la présente décision les modalités de prise en compte de la durée des pratiques.
S’agissant de Novartis
1272.Au cas présent, comme cela ressort du paragraphe 1072 de la présente décision, l’infraction imputée à Novartis a débuté le 9 mai 2011 pour prendre fin au début du mois de novembre 2013.
1273.En conséquence, la durée des pratiques étant de 2 ans et 5 mois, il convient de retenir un coefficient multiplicateur de 1,70.
S’agissant de Roche / Genentech
1274.D’une part, s’agissant des pratiques imputées à Roche, comme cela ressort du paragraphe 1080 de la présente décision, l’infraction a débuté le 7 avril 2008 pour prendre fin au début du mois de novembre 2013. Ainsi, la durée des pratiques de cette société étant de 5 ans et 6 mois, il convient de retenir un coefficient multiplicateur de 3,25.
1275.D’autre part, s’agissant des pratiques imputées à Genentech, comme cela ressort du paragraphe 1086 de la présente décision, l’infraction a débuté le 28 avril 2011 pour prendre fin au début du mois de novembre 2013, soit une durée de 2 ans et 6 mois.
1276.Or, comme indiqué aux paragraphes 1208 à 1210 ci-dessus, la sanction pécuniaire prononcée à l’encontre du groupe Roche est assise sur une seule valeur des ventes, justifiant le prononcé d’une sanction unique pour l’ensemble des sociétés du groupe Roche.
1277.Dès lors, dans la mesure où la durée de participation de Genentech est inférieure à celle de Roche, il conviendra de tenir Genentech solidaire de cette sanction pécuniaire à hauteur de sa durée de participation, correspondant à 45 % de la durée de participation de Roche.
Conclusion sur la détermination du montant de base
1278.Eu égard à la gravité des faits et au dommage causé à l’économie par les pratiques en cause, le montant de base des sanctions pécuniaires, déterminé en proportion des ventes en relation avec l’infraction réalisées par les entreprises concernées, d’une part, et en fonction de la durée de l’infraction, d’autre part, est le suivant :
| Sociétés | Montant de base (en euros) |
| Novartis Pharma SAS et Novartis AG | 69 972 000 |
| Roche | 35 146 310 |
b) Sur l’individualisation des sanctions
En ce qui concerne les circonstances aggravantes et atténuantes
1279.L’Autorité a rappelé aux paragraphes 1174 et 1175 de la présente décision les modalités de prise en compte des éventuelles circonstances aggravantes ou atténuantes.
1280.Les sociétés mises en cause soutiennent que les sanctions pécuniaires prononcées à leur encontre devraient être revues à la baisse compte tenu de l’existence de circonstances atténuantes. Plus spécifiquement, elles font valoir le contexte particulier dans lequel les pratiques se sont déroulées, ainsi que le caractère inédit des infractions sanctionnées, dont la qualification est complexe et ambigüe, justifieraient qu’aucune sanction pécuniaire ne lui soit infligée ou, à tout le moins, le prononcé d’une amende symbolique.
1281.Toutefois, en premier lieu, comme souligné au paragraphe 1228 de la présente décision, les pratiques sanctionnées au titre du grief n° 2 ne revêtent aucun caractère inédit.
1282.En second lieu, pour les raisons invoquées au paragraphe 1178 de la présente décision, si les pratiques en cause se sont effectivement inscrites dans un contexte marqué par un débat scientifique sur l’efficacité et la sécurité d’utilisation d’Avastin « hors AMM » en ophtalmologie, cela ne justifie pas, en l’espèce, une quelconque réduction de sanction, dans la mesure où le discours de Novartis et de Roche, avec l’aide de Genentech, ne s’est pas limité à une présentation objective du contexte d’incertitude scientifique.
1283.Par ailleurs, Roche estime que sa sanction devrait être modérée, compte tenu du fait que sa participation à l’infraction sanctionnée au titre du grief n° 2 est limitée à quelques échanges avec l’autorité de santé et qu’elle n’aurait pas été moteur de l’infraction, contrairement à Novartis.
1284.Toutefois, les contacts de Roche avec les autorités publiques ne se sont pas limités aux échanges avec l’ANSM, puisque le laboratoire a également dirigé son discours vers la ministre de la santé ou la DGS (cf. paragraphes 491 à 494 de la présente décision). Par ailleurs, Roche ne présente aucun élément qui serait de nature à démontrer que Novartis serait à l’origine des pratiques, en aurait été le moteur et/ou que Novartis aurait pris des mesures pour contraindre Roche et Genentech à participer à l’infraction.
1285.Il ressort de ce qui précède qu’aucune circonstance atténuante ne justifie de revoir le montant des sanctions prononcées à la baisse. Par ailleurs, aucun élément du dossier ne permet de considérer qu’une des entreprises sanctionnées devrait voir le montant de sa sanction augmenté au titre de circonstances aggravantes.
En ce qui concerne les circonstances aggravantes et atténuantes
1286.L’Autorité a rappelé aux paragraphes 1180 à 1185 de la présente décision les modalités de prise en compte de la puissance économique.
Sur la puissance économique du groupe Novartis
1287.L’infraction sanctionnée au titre du grief n° 2 a été imputée aux sociétés Novartis Pharma SAS et Novartis AG, en tant qu’auteures, et aux sociétés Novartis Groupe France SA et Novartis AG, en tant que sociétés mères.
1288.Or, comme souligné au paragraphe 1187 de la présente décision, ces sociétés appartiennent au groupe Novartis, qui jouit d’une taille et d’une puissance économique importantes.
1289.La valeur des ventes retenue comme assiette de la sanction prononcée à son égard au titre du grief n° 2 représente ainsi seulement 0,69 % du chiffre d’affaires total du groupe et environ 2,8 % du résultat net consolidé.
1290.Compte tenu de ces éléments, le montant de base de la sanction pécuniaire infligée aux sociétés Novartis Pharma SAS et Novartis AG, solidairement avec leurs sociétés mères, Novartis Groupe France SA et Novartis AG, doit être augmenté de 50 %.
1291.Eu égard à l’ensemble des éléments qui précèdent, le montant de la sanction pécuniaire à imposer aux sociétés Novartis Pharma SAS et Novartis AG, conjointement et solidairement avec les sociétés Novartis Groupe France SA et Novartis AG, sera fixé à 104 958 000 euros au titre du grief n°2.
Sur la puissance économique du groupe Roche
1292.L’infraction sanctionnée au titre du grief n° 2 a été imputée aux sociétés Roche et Genentech Inc., en tant qu’auteures, et à la société Roche Holding AG, en tant que société mère de ces deux sociétés.
1293.Or, ces sociétés appartiennent au groupe Roche, qui jouit d’une taille et d’une puissance économique importantes. Les ressources financières globales du groupe Roche sont en effet très élevées. Son chiffre d’affaires consolidé hors taxes s’élève à 63 751 millions CHF au 31 décembre 2019, soit environ 57 325 millions d’euros(56). Son résultat net consolidé s’établit à 14 108 millions CHF la même année, soit 12 686 millions d’euros.
1294.La valeur des ventes retenue comme assiette de la sanction prononcée à son égard représente ainsi seulement 0,13 % du chiffre d’affaires total du groupe et environ 0,61 % du résultat net consolidé.
1295.Compte tenu de ces éléments, le montant de base des sanctions pécuniaires infligées aux sociétés Roche et Genentech Inc., solidairement avec leur société mère Roche Holding AG, doit être augmenté de 70 %.
1296.Eu égard à l’ensemble des éléments qui précèdent, le montant des sanctions à imposer à la société Roche, conjointement et solidairement avec la société Roche Holding AG, sera fixé à 59 748 726 euros, dont 26 886 927 euros conjointement et solidairement avec la société Genentech Inc.
c) Sur la réitération
1297.L’Autorité a rappelé aux paragraphes 1191 à 1195 de la présente décision les modalités de prise en compte de la situation de réitération.
1298.Pour les raisons évoquées aux paragraphes 1196 à 1201 de la présente décision, il y a lieu de constater que la société Novartis Pharma SAS se trouve dans une situation de réitération, ce qui du reste, n’est pas contesté par Novartis.
1299.En conséquence, il convient de retenir, dans les circonstances de l’espèce, une majoration de 25 % de la sanction prononcée à l’encontre de Novartis Pharma, portant celle-ci à un montant de 131 197 500 euros au titre du grief n° 2.
1300.De même, pour les raisons évoquées aux paragraphes 1199 et 1200 de la présente décision, il y a lieu de constater que Novartis AG et Novartis Groupe France SA ne se trouvent pas en situation de réitération.
1301.Il résulte de ce qui précède que (i) la sanction prononcée à l’encontre de Novartis au titre du grief n° 2 sera de 131 197 500 euros et (ii) Novartis Pharma SAS sera tenue seule responsable de la majoration de la sanction prononcée au titre de la réitération, correspondant au montant suivant : 26 239 500 euros.
d) Conclusion sur le montant de la sanction pécuniaire
1302.Au vu des considérations qui précèdent, il y a lieu d’imposer les sanctions suivantes au titre du grief n° 2 :
- concernant Novartis : une sanction pécuniaire de 131 197 500 euros, étant précisé que (i) Novartis Pharma SAS sera tenue seule responsable de cette sanction à hauteur de 26 239 500 euros(57), et (ii) Novartis Pharma SAS, Novartis AG et Novartis Groupe France SA seront tenues solidairement et conjointement responsables de cette sanction à hauteur de 104 958 000 euros.
- concernant Roche / Genentech : une sanction pécuniaire de 59 748 726 euros, étant précisé que (i) Genentech sera tenue responsable de cette sanction, en tant qu’auteur, à hauteur de 26 886 927 euros(58), et (ii) Roche Holding AG sera tenue, en tant que société mère de Roche et Genentech, solidairement et conjointement responsable de la totalité de la sanction prononcée au titre du grief n° 2.
e) Sur le maximum légal
1303.L’Autorité a rappelé au paragraphe 1203 de la présente décision les conditions d’appréciation du maximum légal.
Concernant le groupe Novartis
1304.Pour les raisons évoquées au paragraphe 1204 de la présente décision, il n’y a pas lieu de modifier le montant de la sanction pécuniaire retenu au titre du grief n° 2.
Concernant le groupe Roche
1305.Le chiffre d’affaires mondial consolidé hors taxes le plus élevé connu réalisé par le groupe Roche était de 63 751 millions CHF au 31 décembre 2019 euros, soit environ 57 325 millions d’euros(59).
1306.Le montant de sanction retenu précédemment étant inférieur à 10 % de ce chiffre, il n’y a pas lieu de le modifier.
DÉCISION
Article 1er : Il est établi que les sociétés Novartis Pharma SAS et Novartis AG, en tant qu’auteures des pratiques, et les sociétés Novartis Groupe France SA et Novartis AG, en leurs qualités de sociétés mères, ont enfreint les dispositions de l’article L. 420-2 du code de commerce ainsi que celles de l’article 102 du traité sur le fonctionnement de l’Union européenne, en mettant en œuvre une pratique de dénigrement d’Avastin sur les marchés français du traitement de la DMLA exsudative par anti-VEGF et des autres indications oculaires (OMD, OBVR, OVCR ou baisse visuelle due à une NVC) traitées par anti-VEGF, combinant la ville et l’hôpital.
Article 2 : Il est établi que les sociétés Novartis Pharma SAS, Novartis AG, Roche SAS et Genentech Inc., en tant qu’auteures des pratiques, et les sociétés Novartis Groupe France SA, Novartis AG et Roche Holding AG, en leurs qualités de sociétés mères, ont enfreint les dispositions de l’article L. 420-2 du code de commerce ainsi que celles de l’article 102 du traité sur le fonctionnement de l’Union européenne, en diffusant un discours alarmiste, voire trompeur, auprès des autorités publiques sur les risques liés à l’utilisation d’Avastin sur les marchés français du traitement de la DMLA exsudative par anti-VEGF et des autres indications oculaires (OMD, OBVR, OVCR ou baisse visuelle due à une NVC) traitées par anti-VEGF, combinant la ville et l’hôpital.
Article 3 : Au titre de la pratique visée à l’article 1er, il est infligé conjointement et solidairement aux sociétés Novartis Pharma SAS, Novartis AG et Novartis Groupe France SA, la sanction pécuniaire de 253 905 750 euros, dans les conditions exposées au paragraphe 1202 de la présente décision.
Article 4 : Sont infligées, au titre de la pratique visée à l’article 2, les sanctions pécuniaires suivantes :
- 131 197 500 euros, conjointement et solidairement aux sociétés Novartis Pharma SAS, Novartis AG et Novartis Groupe France SA dans les conditions exposées au paragraphe 1302 de la présente décision ;
- 59 748 726 euros, conjointement et solidairement aux sociétés Roche, Genentech Inc., et Roche Holding AG, dans les conditions exposées au paragraphe 1302 de la présente décision.
NOTES :
1 Ce résumé a un caractère strictement indicatif. Seuls font foi les motifs de la décision numérotés ci-après.
2 Ordonnance avant-dire droit de la cour d’appel de Versailles du 19 février 2015 (désistement de Roche) ; ordonnance de la cour d’appel de Versailles du 7 mai 2015 (annulant la saisie des correspondances et messages sans lien avec les pratiques ou couvertes par le secret des correspondances avec un avocat) et arrêt de la Cour de cassation du 26 octobre 2016 (rejetant le pourvoi de Novartis).
3 Appelée également « forme pharmaceutique » : forme selon laquelle un traitement est administré.
4 Rapport de synthèse des « Assises du médicament », 23 juin 2011, repris dans l’étude d’impact du projet de loi relatif au renforcement de la sécurité sanitaire du médicament et des produits de santé, juillet 2011.
5 Décret n° 94-568 du 8 juillet 1994 relatif aux autorisations temporaires d'utilisation de certains médicaments à usage humain et modifiant le code de la santé publique.
6 Décret n° 2005-1023 du 24 août 2005 relatif au contrat de bon usage des médicaments et des produits et prestations mentionné à l'article L. 162-22-7 du code de la sécurité sociale et décret n° 2008-1121 du 31 octobre 2008 relatif au contrat de bon usage des médicaments et des produits et prestations mentionné à l'article L. 162-22-7 du code de la sécurité sociale.
7 Le Comité économique des produits de santé (ci-après, « CEPS ») est un organisme interministériel placé sous l’autorité conjointe des ministres chargés de la santé, de la sécurité sociale et de l’économie, est principalement chargé par la loi de fixer les prix des médicaments et les tarifs des dispositifs médicaux à usage individuel pris en charge par l’assurance maladie obligatoire.
8 Le prix est fixé en référence aux prix dans les autres pays européens, plus particulièrement l’Espagne, l’Italie, l’Allemagne et le Royaume-Uni.
9 C'est-à-dire un médicament réservé aux patients hospitalisés.
10 https://ansm.sante.fr/content/download/78441/994175/version/1/file/RTU_Avastin-DMLA_Decision_24-06-2015.pdf
11https://www.ansm.sante.fr/var/ansm_site/storage/original/application/49c1beec1315276b8d7c5e314d362ce5.pdf
12 http://circulaires.legifrance.gouv.fr/pdf/2016/05/cir_40950.pdf
13 https://www.has-sante.fr/portail/upload/docs/evamed/CT-16200_DMLA_PIC_REEV_Avis3_CT16200&16091&16196.pdf
14https://www.legifrance.gouv.fr/affichTexte.do?cidTexte=JORFTEXT000028134502&fastPos=14&fastReqId=1844383847&categorieLien=id&oldAction=rechTexte
15 https://www.vidal.fr/Medicament/eylea_40_mg_ml_sol_inj_en_flacon-124108-indications.htm
16 C'est-à-dire une association de deux ou plusieurs maladies.
17 C’est-à-dire affectant la totalité de l’organisme, non localisés dans l’œil.
18 Le critère d’efficacité dans les études de comparaison Avastin/Lucentis était lié au nombre de lettres pouvant être lues par les patients dans l’échelle ETDRS.
19 https://ansm.sante.fr/var/ansm_site/storage/original/application/49c1beec1315276b8d7c5e314d362ce5.pdf
20 Le (RCP) est un document destiné aux professionnels de santé. Il est fixé par les autorités lors de l'octroi de l'autorisation de mise sur le marché (AMM) ou de l'enregistrement des médicaments à base de plantes. Il précise : la dénomination du médicament ; la composition qualitative et quantitative ; la forme pharmaceutique ; les données cliniques : indications thérapeutiques, posologie et mode d'administration, contre-indications, mises en garde spéciales et précautions particulières d'emploi, interactions avec d'autres médicaments, grossesse et allaitement, effets sur l'aptitude à conduire et à utiliser des machines, effets indésirables, surdosage ; les propriétés pharmacologiques : propriétés pharmacodynamiques, propriétés pharmacocinétiques, données de sécurité préclinique ; les données pharmaceutiques : liste de excipients, incompatibilité, durée de conservation, précaution particulière de conservation, nature et contenu de l'emballage extérieur, instruction pour l'utilisation, la manipulation et l'élimination ; le titulaire de l'autorisation d'utilisation ; la résentation et le numéro d'identification administrative ; la date de première autorisation/renouvellement de l'autorisation ; et la date de mise à jour du texte. https://ansm.sante.fr/Glossaire/(filter)/R.
21 La pharmacocinétique est l’étude du devenir d’un médicament dans l’organisme.
22 La pharmacodynamique est l’étude de l’effet d’un médicament dans l’organisme.
23 Une étude ou un essai randomisé est un protocole expérimental ayant pour but d’évaluer l’efficacité d’une thérapie, d’une action de prévention ou d’un médicament, et comparant un groupe expérimental testant le traitement concerné à un groupe « contrôle », suivant les recommandations standard ou prenant un placebo.
24 La choroïde est la membrane intermédiaire de l’œil située entre la rétine et la membrane externe.
25 https://solidarites-sante.gouv.fr/fichiers/bo/2015/15-09/ste_20150009_0000_0057.pdf
26 https://www.has-sante.fr/portail/upload/docs/application/pdf/2013-04/description_de_la_regulation_de_la_promotion_des_produits_de_sante_-_2013.pdf
27 Ce manuel a été élaboré dans le cadre d'un projet mené en collaboration entre Action Internationale pour la Santé (HAI) et l'Organisation mondiale de la santé (OMS) ; il est axé sur la promotion. Il a été rendu possible grâce à l'expertise et au savoir de nombreux membres de l'OMS et de HAI et d'un grand groupe d'enseignants et de militants oeuvrant avec le réseau AIS sur la promotion pharmaceutique.
28 http://ansm.sante.fr/var/ansm_site/storage/original/application/a31f2f8418a0ed7bb4e6ff5e25aa7c88.pdf
29 Mission de l’action régionale.
30 Étude qui examine les transformations d’un produit sous certaines conditions (température, humidité, etc.), afin de déterminer la durée de vie et les conditions de stockage du produit.
31 Ce paramètre correspond au temps nécessaire pour que, après l’administration d’un médicament, sa concentration plasmatique diminue de moitié.
32 La cour d’appel de Paris a fait application de ce principe pour juger que le défaut de notification du rapport au ministre n’avait pas été susceptible d’exercer une influence sur le sens de la décision de l’Autorité et n’a pu causer grief aux entreprises mises en cause (arrêt du 11 juillet 2019, société Jansen-Cillag SAS, n° 18/01945).
33 Les publications concernées étaient les suivantes : recommandation temporaire d’utilisation pour Avastin dans le traitement de la DMLA, plusieurs avis concernant l’utilisation de Lucentis pour le traitement de la DMLA, avis sur l’inscription d’Eylea pour le traitement de la DMLA, recommandations de bonne pratique concernant le traitement de la DMLA (prise en charge diagnostique et thérapeutique).
34 Synthèse des travaux sur les « Assises du médicament ».
35 Lignes directrices relatives à la notion d'affectation du commerce figurant aux articles 81 et 82 du traité, Journal officiel n° C 101 du 27/04/2004 p. 0081 – 0096.
36 Le Programme de médicalisation des systèmes d'information (« PMSI ») est un outil de description et de mesure médico-économique de l'activité hospitalière.
37 Le système de classification anatomique, thérapeutique et chimique (en anglais : Anatomical Therapeutic Chemical (« ATC ») Classification System.
38 Les parts de marché issues du sondage sont incluses, dans 95 % des cas, dans la fourchette définie par l’intervalle de confiance.
39 Voir notamment https://www.aao.org/eye-health/diseases/avastin-eylea-lucentis-difference et https://www.allaboutvision.com/conditions/lucentis-vs-avastin.htm.
40 Puisque l’intervalle de confiance à 95 % des parts de marché d’Avastin s’étend en janvier 2008 de 8 à 22 % (250 injecteurs interrogés mais « 100 réponses par vague », cote 13977) et de 0 à 10 % en juin 2012 (80 injecteurs interrogés, cote 4705).
41 http://ir2.flife.de/data/novartis/igb_html/index.php?bericht_id=1000002&index=120&lang=FRA
42 http://www.omedit-idf.fr/wp-content/uploads/2016/11/RBU-ANSM-07.2012.pdf
43 Prévus dans le décret n° 2005-1023 du 24 août 2005 relatif au contrat de bon usage des médicaments et des produits et prestations mentionné à l'article L. 162-22-7 du code de la sécurité sociale et le décret n° 2008-1121 du 31 octobre 2008 relatif au contrat de bon usage des médicaments et des produits et prestations mentionné à l'article L. 162-22-7 du code de la sécurité sociale.
44 https://www.roche.com/about/governance/ec_bod_former/executive_commitee.htm
45 https://www.roche.com/about/business/pharmaceuticals/roche_pharmaceuticals_business_overview.htm
46 https://www.gene.com/about-us.
47 Travaux préparatoires sur le projet de loi de financement de la sécurité sociale pour l’année 2013, voir en particulier, les propos de Mme Marisol Touraine, ministre de la santé, lors des débats tenus à l’assemblée nationale le 26 octobre 2012 sur l’article 45.
48 Décision n° 20-D-04 du 16 mars 2020 relative à des pratiques mises en œuvre dans le secteur de la distribution de produits de marque Apple, paragraphes 1215 ; Décision n° 18-D-26 du 20 décembre 2018 relative à des pratiques mises en œuvre dans le secteur de la commercialisation de fertilisants liquides pour la production hors-sol dédiés à la culture domestique, paragraphes 339 et 340 ; décision n° 16-D-17 du 21 juillet 2016 relative à des pratiques mises en œuvre dans le secteur des appareils de chauffage mobiles à combustible liquide, paragraphes 180 et 181.
49 Voir l’avis de la Commission de la transparence de la HAS du 19 septembre 2018.
50 Entre le mois d’avril 2008 et le mois de novembre 2013, le nombre mensuel moyen d’injections de Lucentis est environ 25 000 (source : données GERS). Par ailleurs, l’écart de coût moyen entre Lucentis et Avastin sur la même période est d’environ 930 euros.
51 Soit une économie d’environ 9,6 euros par mois par rapport à un prix moyen de Lucentis de l’ordre de 960 euros sur la période allant de mars 2008 à novembre 2013.
52 Sur la base du taux de change moyen BCE observé entre le 1er janvier 2019 et le 31 décembre 2019 (0,8934).
53 Sur la base du taux de change moyen BCE observé entre le 1er janvier 2019 et le 31 décembre 2019 (0,8934).
54 Dans les conditions définies aux paragraphes 1274 à 1277 de la présente décision.
55 Décomposé de la façon suivante : […] euros, s’agissant des redevances perçues par Novartis, d’une part, et […] euros, s’agissant des ventes de principe actif de Lucentis. Une partie de la valeur des ventes communiquée par Roche est en dollars US. Pour celle-ci, le taux de change moyen BCE entre le 1er janvier 2012 et le 31 décembre 2012 (0,7789) est appliqué.
56 Sur la base du taux de change moyen BCE observé entre le 1er janvier 2019 et le 31 décembre 2019 (0,8992).
57 Compte tenu du constat d’une situation de réitération pour la seule société Novartis Pharma SAS (cf. paragraphes 1297 à 1301 de la présente décision).
58 Compte tenu de la durée de participation inférieure de Genentech à l’infraction (cf. paragraphes 1276 et 1277 de la présente décision).
59 Sur la base du taux de change moyen BCE observé entre le 1er janvier 2019 et le 31 décembre 2019 (0,8992).